Nat. Commun. | Ingemar André团队开发复杂对称蛋白质组装体预测方法EvoDock

Nat. Commun. | Ingemar André团队开发复杂对称蛋白质组装体预测方法EvoDock

DrugOne
发布于 2026-03-25 14:37:06
发布于 2026-03-25 14:37:06
2023年12月16日,瑞典隆德大学的Ingemar André团队在Nature Communications上发表了一篇题为《Accurate prediction of protein assembly structure by combining AlphaFold and symmetrical docking》的论文。这篇文章介绍了一种结合AlphaFold预测与对称分子对接的新方法EvoDOCK,用于高精度预测具有立方对称性的大型蛋白质组装体结构。该方法通过亚基构象库生成、对称性参数降维和进化算法优化,成功实现了对四面体、八面体和二十面体等复杂对称系统的结构组装。测试结果显示,在27个立方对称系统中,局部组装与全局组装的中位TM-score均达到0.99,多数模型达到接近实验解析精度。该研究突破了AlphaFold在处理多链大复合体时的限制,为病毒衣壳、人工蛋白质笼等复杂结构的预测与设计提供了新途径。

背景
蛋白质是生命活动的执行者,而许多关键功能(如DNA复制、能量合成)依赖于多个蛋白质亚基形成的复杂组装结构。理解这些组装体的三维结构,是揭示其功能机制的核心。近年来,深度学习技术AlphaFold的横空出世,彻底改变了蛋白质结构预测领域——它能以原子级精度预测单体和简单多聚体的结构。然而,当面对由数十甚至上百个亚基组成、具有高度对称性的大型蛋白质复合体(如病毒衣壳)时,AlphaFold却显得力不从心。
这类大型组装体通常呈现立方对称性(如四面体、八面体、二十面体),其亚基排列紧密,界面相互作用复杂。传统实验方法(如冷冻电镜)耗时耗力,而现有计算工具因计算资源限制和采样效率低下,难以处理如此庞大的系统。针对这个情况,瑞典隆德大学的Ingemar André团队在Nature Communications发表研究,提出了一种结合AlphaFold预测与对称性引导的分子对接的新方法EvoDock,成功实现了立方对称蛋白质组装体的高精度结构预测。该方法不仅填补了AlphaFold的空白,更为研究病毒、分子机器等复杂系统提供了全新工具。
方法
研究团队将巨型蛋白质组装的预测分解为三大模块,通过“亚基预测-对称约束-全局优化”的协同策略(如图1所示)攻克技术难关,具体步骤如下。
(1)亚基预测与动态修剪:针对目标蛋白质的每一个亚基,分别运行AlphaFold和AlphaFold-Multimer多次(例如100次),构建包含不同构象的亚基结构库。随后,依据pLDDT(局部结构置信度,>90为高置信)和0.8·ipTM + 0.2·pTM(AFM界面预测评分,>0.9为高质量界面)等标准对生成的模型进行筛选。由于AlphaFold对N/C端残基的预测误差通常较大,团队还开发了一套自动化动态修剪策略,包括计算每个残基的pLDDT均值、二级结构倾向性(通过DSSP算法)以及邻近残基的接触频率,并从两端逐步去除残基,直至所有指标均达到预设阈值(pLDDT大于等于90,二级结构倾向性大于等于70%,接触频率大于等于70%)。最后,通过剔除结构高度相似(RMSD<0.1 Å)的模型,保留构象多样性,构建出高质量的亚基构象库。
(2)对称约束:在对称框架构建与参数降维方面,研究团队利用立方对称性(如图1b的T、O、I三类)将系统的自由度从数百万大幅压缩至六个关键参数:三个旋转参数(ψ、θ、φ分别对应绕x、y、z轴的旋转)和三个平移参数(z表示组装体半径,x为寡聚体到原点的距离,λ为整体旋转角)。为避免结构崩溃,团队引入数学约束机制,例如将λ的旋转范围限制为360°/n(n为对称轴阶数,如二十面体的5轴对应λ∈[-72°,72°])。此外,根据是否已知模板信息,采用双模式采样策略:在局部组装中,参数在实验值附近小范围扰动;而在全局组装中,部分参数(如z)从AFM预测的寡聚体中提取,其余参数则在宽范围内随机采样,以覆盖更广的构象空间。
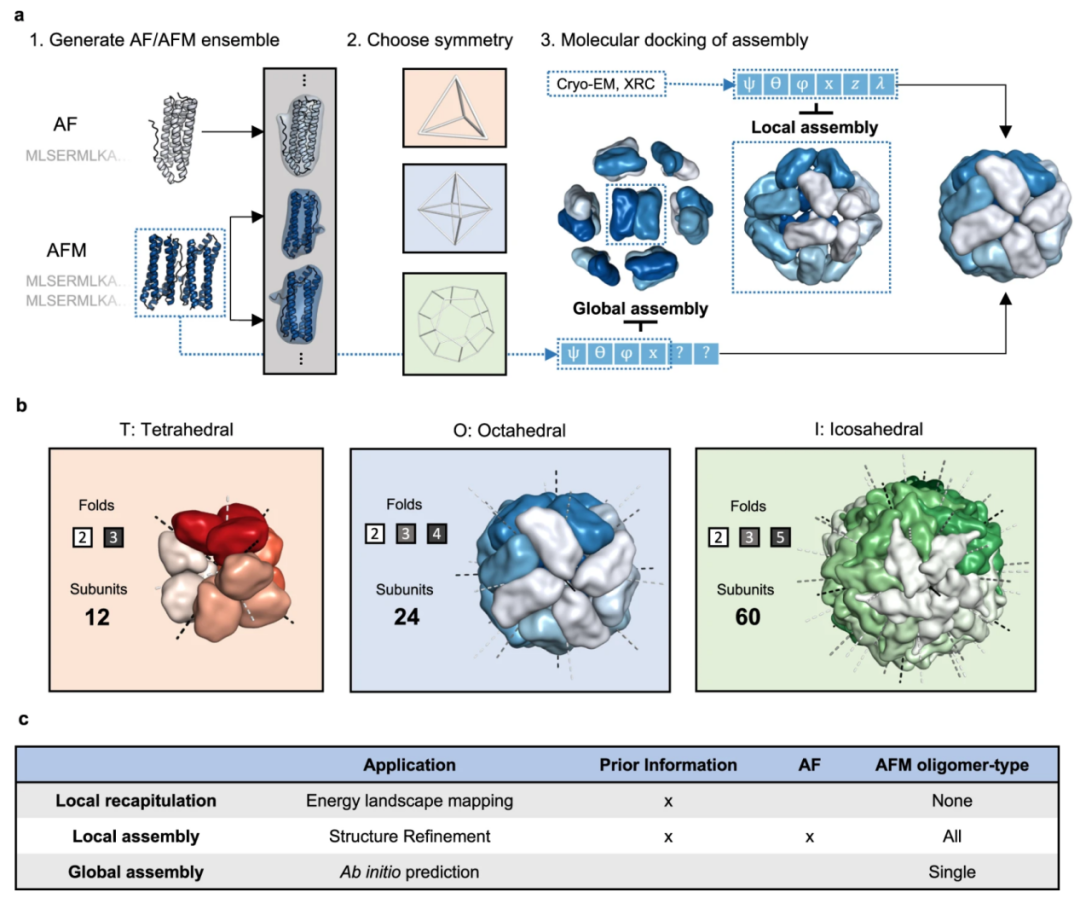
图1. 组装预测方法及立方对称群示意图。
(3)全局优化:研究团队进一步基于其先前开发的EvoDOCK算法(原用于异源二聚体对接),针对对称组装系统进行了四项关键升级,形成“对称EvoDOCK”流程(如图2所示)。该流程采用差分进化与蒙特卡洛相结合的混合优化方法:首先初始化种群,每个个体包含随机选取的亚基构象及六个对称参数;随后在进化阶段通过突变和重组推动全局搜索;并辅以Rosetta全原子能量函数(REF2015)进行局部精细优化。为有效避免空间冲突,团队设计了“滑动移动”策略,使亚基沿对称轴以0.3Å为步长逐步移动,最多尝试100步,并优先处理高阶对称轴(如二十面体的5轴)。同时,开发了CloudContactScore(CCS)快速碰撞检测函数,通过点云简化(仅保留骨架原子N、C、O、CA、CB)并综合四项评分(碰撞惩罚、氢键奖励、疏水接触等),将检测效率提升了10倍。最终,对筛选出的Top 100模型进行Rosetta全原子松弛和k-means聚类,每类选取能量最低的代表模型作为最终预测结果。
针对AlphaFold-Multimer(AFM)在预测全部界面时的局限性(例如立方系统需三种界面而AFM仅能预测其中一种),研究团队提出了局部补全策略:保留AFM已预测的正确界面,利用EvoDOCK搜索其余界面的最优排布方式。同时,为应对可能的180°翻转对称性,引入双取向采样方法,同步运行两种初始取向的模拟,最终依据能量高低选择最优方向。
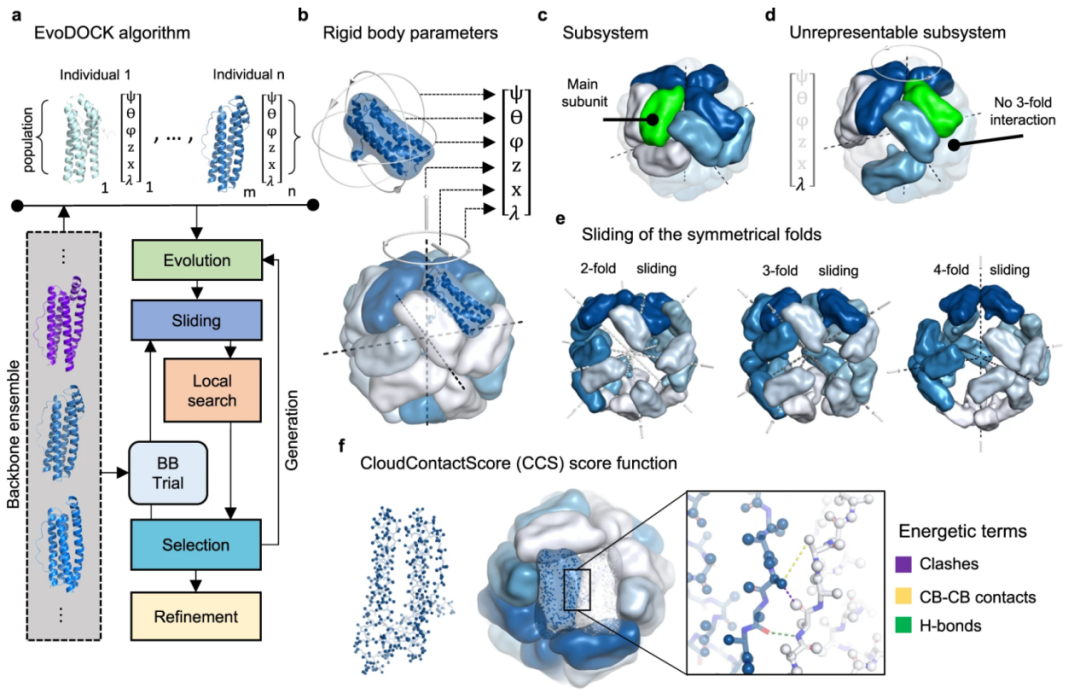
图2. 对称性EvoDOCK方法。
结果
研究团队选取了27个具有明确立方对称性的蛋白质复合体(包括9个四面体、9个八面体和9个二十面体),涵盖病毒衣壳与分子伴侣等多种功能类型,如图3所示。测试结果表明,在局部组装实验中(已知亚基结构),预测模型的中位TM-score高达0.99(1.0为完美匹配),DockQ评分为0.76,RMSD仅为1.2Å;所有模型均达到“可接受”质量(TM-score≥0.9,DockQ≥0.23),其中59%更达到“高质量”(DockQ≥0.8)。在更具挑战的全局组装实验中(仅凭序列信息),中位TM-score仍保持在0.99,DockQ评分为0.72,成功预测率达到89%。典型案例如二十面体病毒衣壳5EKW,其预测结构与实验模型几乎完全重叠。这一方法实现了从“不可能”到“高精度”的突破,传统方法因计算复杂度难以处理超过10条链的组装体,而本研究成功预测了含60个亚基的二十面体系统,例如噬菌体衣壳蛋白(PDB: 1X36)的全局组装RMSD仅1.5Å,DockQ评分达0.85,接近实验解析精度。
进一步的普适性验证显示,在111个测试序列中,78%的单体结构可通过AlphaFold准确预测(pLDDT≥90),72%的界面可通过AFM预测(0.8·ipTM + 0.2·pTM≥0.9)。综合两者,57%的立方系统满足高质量输入要求,且这一比例预计将随AlphaFold的持续迭代而进一步提升。
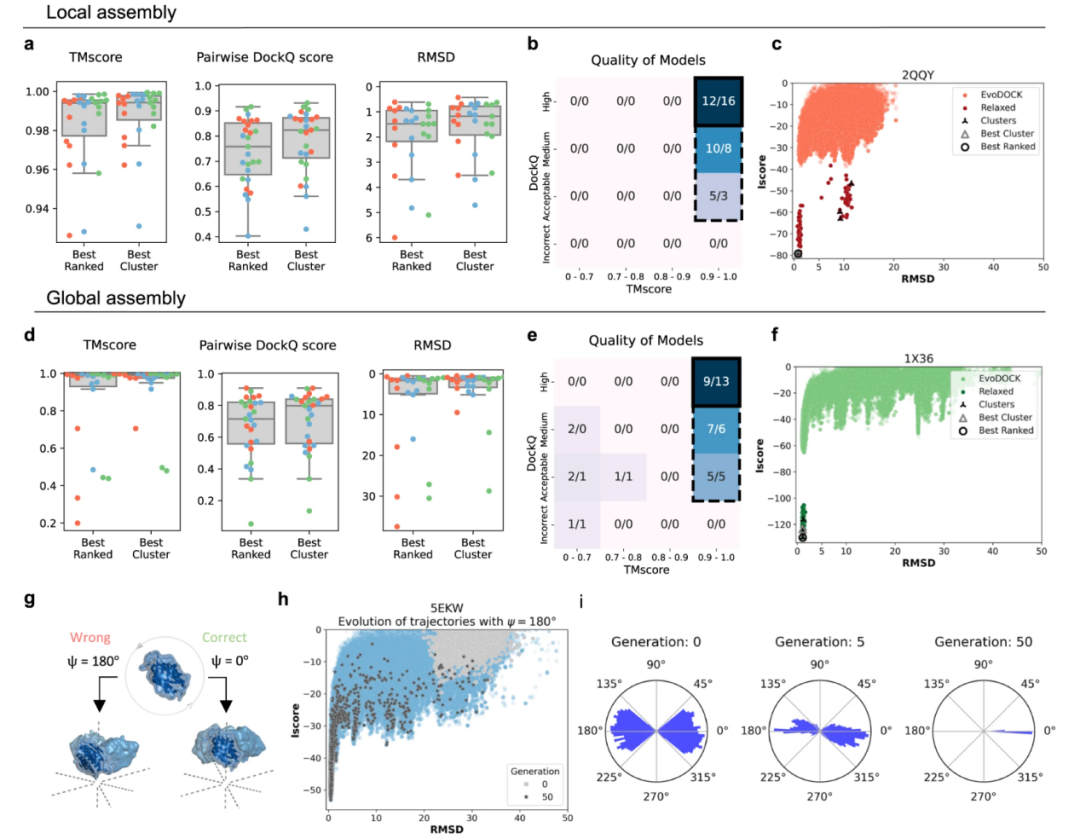
图3. 局部与全局组装实验结果。
讨论
该研究不仅突破了大型对称组装体的预测极限,也为病毒学研究、纳米生物技术及疾病机制解析开辟了新路径。例如,在病毒学领域,可快速解析未知病毒衣壳结构,助力疫苗与抗病毒药物研发;在纳米生物技术中,有助于设计人工蛋白质笼状结构,用于药物递送或生物传感;同时,该方法也能用于揭示癌症相关蛋白复合体的异常组装机制。未来,团队计划将方法扩展至准对称系统(如复杂病毒衣壳)和异源多聚体,并整合冷冻电镜密度图以进一步提升预测精度。
总之,AlphaFold的诞生被誉为“结构生物学的革命”,而这项研究将其边界推向了更高维度的复杂性。随着深度学习与物理驱动的分子对接方法的深度融合,我们离“从序列到功能”的终极目标更近一步。
文章链接:
https://www.nature.com/articles/s41467-023-43681-6
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号