【ChiP-seq分析】超级增强子系列7: 用MEME进行超级增强子转录因子motif 富集分析实战

【ChiP-seq分析】超级增强子系列7: 用MEME进行超级增强子转录因子motif 富集分析实战

三兔测序学社
发布于 2025-12-29 12:55:17
发布于 2025-12-29 12:55:17
一、引言:为什么要做超级增强子的motif分析?
超级增强子(Super Enhancers, SEs)是一类具有高度转录活性的调控区域,通常富集大量转录因子结合位点,驱动关键基因(如发育调控基因、癌基因)的表达。解析超级增强子中富集的转录因子结合motif,是揭示其调控机制、识别关键调控因子的核心步骤。
本篇博文将带你完整走一遍超级增强子motif分析流程,重点介绍如何使用 bedtools getfasta 提取SE区域序列,并推荐使用 MEME在线工具中的XSTREME 进行denovo motif发现。同时,我们将深入对比 STREME、XSTREME、MEME-ChIP、GLAM2、MoMo 等主流工具的相同点与差异点,并阐明为何XSTREME是分析超级增强子的理想选择。
二、第一步:从SE.bed到SE.fasta——使用bedtools getfasta提取序列
在进行motif分析前,必须将基因组坐标形式的 SE.bed 文件转换为包含实际DNA序列的 SE.fasta 文件。这一步我们使用强大的 bedtools 工具包中的 getfasta 命令。
1. 准备工作
全基因组参考序列文件(如 hg38.fa)
超级增强子区域的BED文件(SE.bed),格式示例如下:
2. 使用bedtools getfasta提取序列
bedtools getfasta \
-fi hg38.fa \
-bed SE.bed \
-fo SE.fasta \
-name \
-s
#参数解释:
-fi: 输入的参考基因组fasta文件
-bed: 输入的BED格式区域文件
-fo: 输出的fasta文件
-name: 使用BED文件第4列(name字段)作为FASTA序列的header,便于后续追踪
-s: 考虑链方向,若peak在负链,则输出其反向互补序列最终你会得到一个如下的FASTA文件:
>SE1::chr1:1000000-1050000(+)
AGCTAGCTAGCT...
>SE2::chr2:2000000-2100000(-)
TCGATCGATCGA...这个 SE.fasta 文件即可作为输入,上传至MEME suite进行下一步分析。
三、进入MEME在线平台:选择XSTREME进行motif发现
1. 访问 http://meme-suite.org/index.html,选择 "Motif Discovery" 模块,你会看到多个工具可供选择。我们重点关注以下五个:
工具名称 | 核心功能 | 适用序列类型 | 关键特性/优势 |
|---|---|---|---|
MEME | 无间隔基序发现 | DNA, RNA, 蛋白质 | 经典算法,寻找保守的无间隔序列模式 |
STREME | 无间隔基序发现 | DNA, RNA | 速度快,适合大规模数据集,支持对照组比较 |
GLAM2 | 有间隔基序发现 | DNA, 蛋白质 | 允许插入和删除,适合寻找结构更复杂的基序 |
MEME-ChIP | ChIP-seq综合分析 | DNA | 专为ChIP-seq数据设计,整合了多种分析算法 |
XSTREME | 全序列综合分析 | DNA | 适用于基序位置不固定的全序列分析 |
AME | 基序富集分析 | DNA, RNA | 测量已知基序在序列集中的富集程度 |
CentriMo | 中心富集分析 | DNA | 专门检测基序是否集中在序列中心区域 |
MoMo | 蛋白质修饰分析 | 蛋白质 | 用于寻找翻译后修饰(PTM)的基序 |
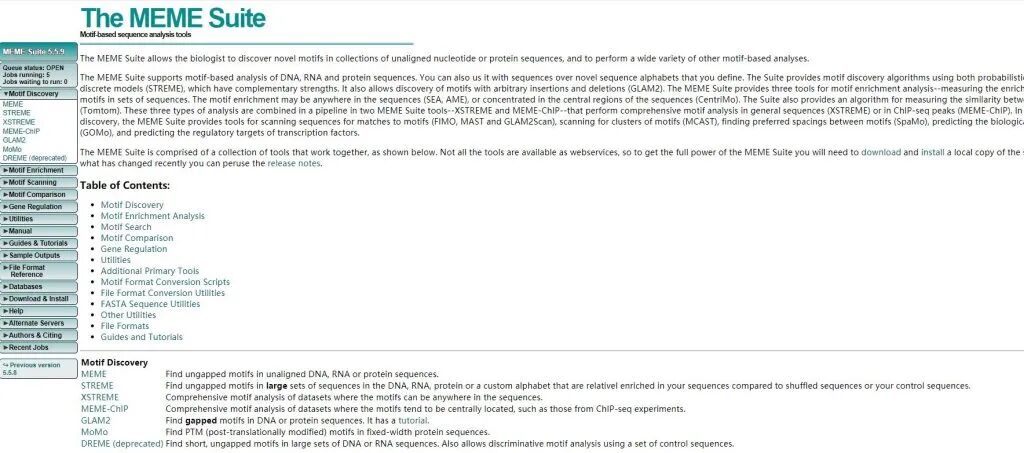
2.进入XSTREME模块,在Input the sequences ,上传Fasta文件。在Input Job details 处填写邮件与任务名称。
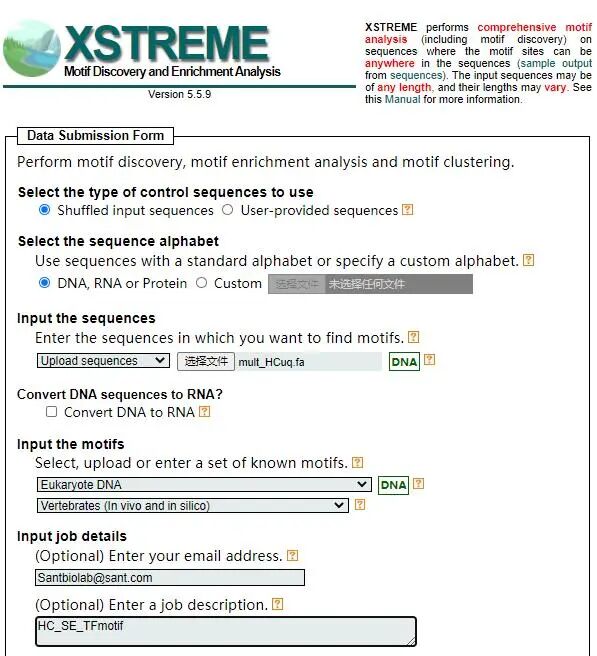
3.填写高级选项并提交任务:通过E-value收紧或者放宽富集条件。在STREME,MEME,SEA options也可以进行相应设置。最后点击Start Search进行motif 分析。
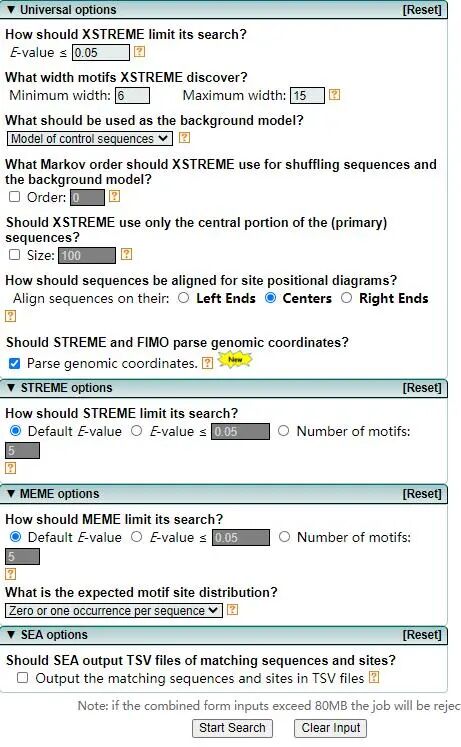
4.查看结果:
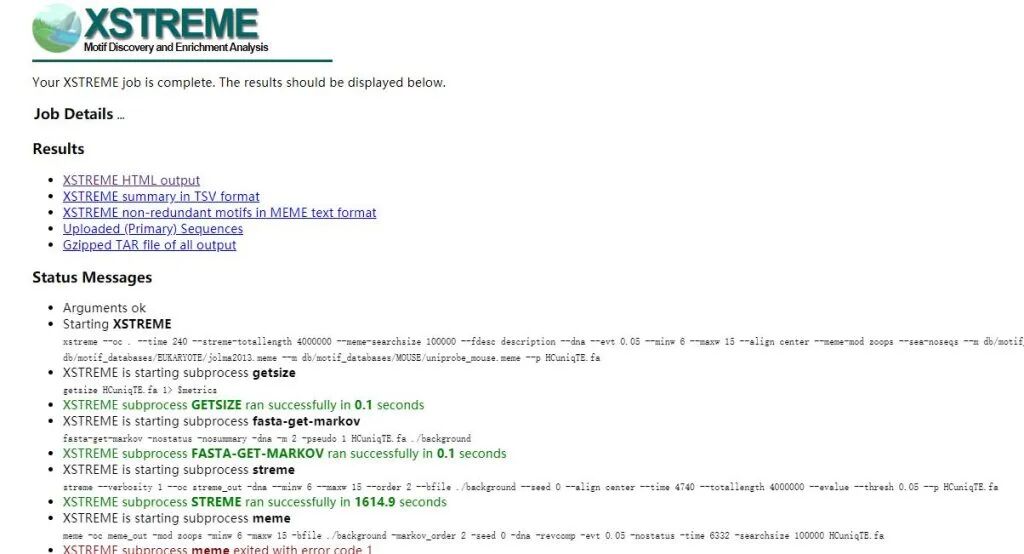
XSTREME HTML output:这是完整的可视化报告,包含富集分析结果、序列标志图(Sequence Logo)、统计表格及基因-基序关联网络图,适合直接在浏览器中查看和解读。
XSTREME summary in TSV format:这是富集分析结果的摘要表格(制表符分隔),包含显著富集的基序ID、转录因子名称、p值、q值等关键统计信息,适合用Excel或文本编辑器打开,便于快速筛选关键基序。
XSTREME non-redundant motifs in MEME text format:这是非冗余基序的标准化描述文件,以MEME工具专用的文本格式存储每个基序的位置权重矩阵(PWM),可用于后续的基序扫描或与其他工具(如FIMO)联动分析。
Uploaded (Primary) Sequences:这是你原始输入的序列文件,作为分析结果的对照参考,确保分析基于正确的输入数据。
Gzipped TAR file of all output:这是所有结果的压缩包(.tar.gz格式),包含上述所有文件,便于一次性下载和本地备份。
四、不同Motif分析工具深度对比:相同点与不同点
MEME、STREME、XSTREME、GLAM2 属于 deno
vo motif discovery 工具;MEME-ChIP 是 集成分析流程;AME 和 CentriMo 是 富集验证类工具;MoMo 则专注于 蛋白质组学场景。
✅ 不同点(核心差异)——基于功能定位的重新梳理
我们不再以单一维度对比,而是从分析目标、算法策略、数据适应性、应用场景四个层面进行系统性拆解:
1. 分析目标差异:从“发现”到“验证”形成完整链条
Denovo 发现类(XSTREME, STREME, MEME, GLAM2)
目标:在未知motif的前提下,从序列中挖掘潜在模式。
区别:
MEME:经典EM算法,适合小规模、高保守性motif。
STREME/XSTREME:基于k-mer频次统计,速度更快,更适合高通量数据。
GLAM2:允许空位(gaps),适合结构域中存在插入/缺失的复杂motif(如某些锌指蛋白结合位点)。
集成分析类(MEME-ChIP)
目标:模拟完整ChIP-seq分析流程,自动执行“发现 → 扫描 → 富集 → 注释”全流程。
特点:内部调用STREME、FIMO、CentriMo等工具,适合标准ChIP-seq peak分析。
富集验证类(AME, CentriMo)
AME:用于验证已知motif在目标序列中是否显著富集(如:SOX2 motif是否在SE区域富集)。
CentriMo:特别关注motif是否集中在peak中心(典型ChIP-seq信号特征),用于过滤假阳性。
特殊用途类(MoMo)
专用于蛋白质序列中翻译后修饰位点(如磷酸化、乙酰化)的motif建模,不适用于DNA调控分析。
2. 算法策略差异:效率与灵活性的权衡
工具 | 算法基础 | 是否支持gapped | 是否支持背景校正 | 是否支持对照比较 |
|---|---|---|---|---|
MEME | EM + 贪心搜索 | 否 | 是 | 否 |
STREME | k-mer频次 + E值筛选 | 否 | 是 | ✅ 是(case/control) |
XSTREME | 扩展k-mer + 并行优化 | 否 | 是 | ✅ 是 |
GLAM2 | 全局比对 + EM | ✅ 是 | 是 | 否 |
MEME-ChIP | 集成流程(含STREME等) | 否 | 是 | 否 |
3. 数据适应性差异:从“标准peak”到“复杂调控区域”
工具 | 适合数据类型 | 是否适合超级增强子 |
|---|---|---|
MEME | 小规模、保守序列 | ⚠️ 一般 |
STREME | 大规模、短motif富集区域 | ✅ 推荐 |
XSTREME | 超大规模、长序列、复杂调控区 | ✅✅ 强烈推荐 |
GLAM2 | 含结构变异的蛋白/DNA结合位点 | ⚠️ 仅当怀疑有gaps时 |
MEME-ChIP | 标准ChIP-seq peak(~500bp) | ⚠️ 可能因序列过长失败 |
AME | 验证已知motif富集 | ✅ 辅助使用 |
CentriMo | 检测motif中心富集 | ⚠️ SE通常无明确中心 |
MoMo | 蛋白质PTM分析 | ❌ 不适用 |
📌 特别说明:超级增强子通常跨越数千bp,包含多个分散的TFBS簇,motif分布广泛且无固定中心,因此:
MEME-ChIP 和 CentriMo 的“中心富集”假设不成立;
XSTREME 的“全序列扫描”策略更符合SE的生物学特性。
4. 应用场景差异:从“通用发现”到“定制化分析”
MEME:教学级工具,适合初学者理解motif发现原理。
STREME/XSTREME:高通量筛选首选,尤其适合筛选关键调控因子。
GLAM2:适合研究结构复杂的调控元件(如增强子中存在插入序列)。
MEME-ChIP:适合标准ChIP-seq数据分析流程,提供一站式解决方案。
AME:用于验证假设,如“MYC motif是否在SE中富集”。
MoMo:完全属于蛋白质组学领域,与DNA调控无关。
五、为何选择XSTREME分析超级增强子?——三大核心优势
结合上述系统性对比,我们再次强调 XSTREME 是分析超级增强子motif的最优选择,理由如下:
✅ 优势一:专为“大规模、长序列”优化,完美匹配SE特征
超级增强子平均长度可达5–10kb,传统工具(如MEME-ChIP)易因内存溢出而失败。XSTREME采用分布式k-mer扫描 + 并行计算架构,能高效处理数千个长序列,确保分析稳定性。
✅ 优势二:高灵敏度捕捉“短而密集”的TFBS簇
SE中常存在多个短motif(如ETS、AP-1、bZIP等)密集排列。XSTREME通过滑动窗口 + 统计显著性筛选,能更灵敏地识别这些“微小但关键”的调控信号,避免被背景噪声掩盖。
✅ 优势三:支持对照比较,提升结果可信度
可上传对照组序列(如普通增强子或随机基因组区域),进行case vs control分析。输出结果包含log2(fold change) 和 P值,便于筛选在SE中特异性富集的motif,直接指向关键调控因子。
🧪 科研建议: 可将XSTREME结果导出为PFM矩阵,使用FIMO在全基因组范围内扫描,构建“SE相关motif靶基因集”,进一步进行功能富集分析(GO/KEGG),形成完整研究闭环。
六、总结:工具选择应服务于生物学问题
生物学问题 | 推荐工具 |
|---|---|
“我的SE区域中有哪些未知的保守motif?” | ✅ XSTREME(首选)或 STREME |
“某个已知TF(如OCT4)的motif是否在SE中富集?” | ✅ AME |
“SE中的motif是否集中在peak中心?” | ⚠️ 不推荐(SE无明确中心) |
“SE相关蛋白的磷酸化位点是否有共同motif?” | ✅ MoMo(仅限蛋白质序列) |
“ChIP-seq peak中有哪些denovo motif?” | ✅ MEME-ChIP 或 STREME |
七、结语:让XSTREME点亮你的超级增强子研究
超级增强子是基因调控网络的“指挥中心”,而motif分析是破译其“密码本”的关键一步。在众多工具中,XSTREME凭借其对大数据集的高效处理能力、对短motif的高灵敏度以及友好的输出格式,成为分析超级增强子motif的首选工具。
结合 bedtools getfasta 的精准序列提取与 MEME suite 的强大分析生态,你将能够:发现驱动疾病或发育的关键转录因子;构建调控网络模型;为后续实验(如突变验证、ChIP-qPCR)提供靶点。
🔬 科研不止于工具,更在于思路。选择正确的工具,才能让数据说话。
【超级增强子系列文章】
超级增强子系列1:super enhancer鉴定-ROSE软件的安装与使用
超级增强子系列2:ROSE准备gff文件:peak 信息文件转化为9列gff格式文件R代码
超级增强子系列3:R语言批量处理ROSE文件生成SE与TE.bed文件
超级增强子系列5:用ChIPseeker进行超级增强子基因注释
如果你觉得这篇博文对你有帮助,请点赞、收藏、转发!支持我们持续输出优质内容!
关注“三兔测序学社”,获取更多实用教程与前沿解读。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-12-12,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号