【ChIP-seq分析】超级增强子系列6:GREAT-基因组调控元件专业注释富集工具

【ChIP-seq分析】超级增强子系列6:GREAT-基因组调控元件专业注释富集工具

三兔测序学社
发布于 2025-12-29 10:14:24
发布于 2025-12-29 10:14:24
安装
GREAT(基因组区域注释富集工具)是一种直接对基因组区域进行的功能富集分析。是一种基因顺式调控元件基因注释工具,如增强子、超级增强子、转录因子结合区域等。 该包实现了GREAT算法(本地GREAT分析),同时支持直接与GREAT网络服务交互(在线GREAT分析)。
rGREAT可在Bioconductor上获取
http://bioconductor.org/packages/devel/bioc/html/rGREAT.html
在线GREAT网站:http://great.stanford.edu/public/html/
在线分析网上由于国内网络不稳定。不做推荐。
使用说明可在github上查看:https://jokergoo.github.io/rGREAT/index.html
下载代码如下
#从Bioconductor上获取
if(!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("rGREAT")
#从
GitHub下载:
library(devtools)
install_github("jokergoo/rGREAT")
核心代码
job = submitGreatJob(peaks_gr, species = "hg38",adv_upstream = 500, adv_downstream = 500, adv_span = 1500)
job = submitGreatJob(bed, species = "hg38",rule = "twoClosest", adv_twoDistance = 2000)
job = submitGreatJob(bed, species = "hg38",rule = "oneClosest", adv_oneDistance = 2000)代码解释
species: “hg38”, “hg19”, “mm10”, “mm9” are supported in GREAT version 4.x.x, “hg19”, “mm10”, “mm9”, “danRer7” are supported in GREAT version 3.x.x and
bgChoise: 背景区域。wholeGenome和data。如果此值设置为data,则应指定bg参数。 includeCuratedRegDoms: 是否包含经过人工整理的调控域。
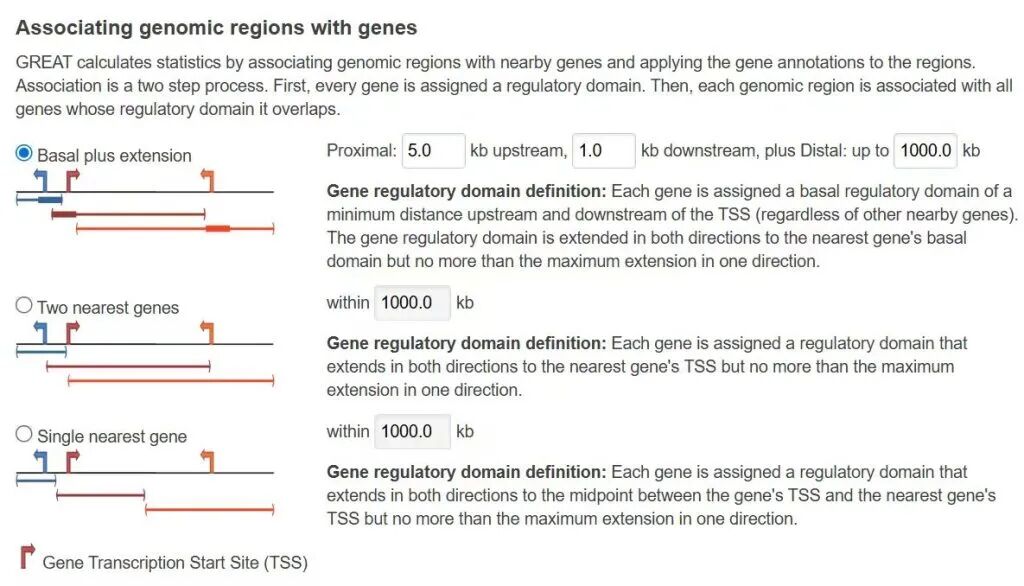
rule: 如何将基因组区域与基因关联。 1.basalPlusExt: 模式“基础域加延伸”。基因调控域定义:每个基因被分配一个基础调控域,包含TSS上游和下游的最小距离(不考虑附近的其他基因)。基因调控域向两个方向延伸到最近基因的基础域,但单向不超过最大延伸距离。 adv_upstream: 上游近端延伸(单位:kb) adv_downstream: 下游近端延伸(单位:kb) adv_span: 最大延伸距离(单位:kb)
2.twoClosest: 模式“两个最近基因”。基因调控域定义:每个基因被分配一个调控域,向两个方向延伸到最近基因的TSS,但单向不超过最大延伸距离。 adv_twoDistance: 最大延伸距离(单位:kb)
3.oneClosest: 模式“单个最近基因”。基因调控域定义:每个基因被分配一个调控域,向两个方向延伸到该基因TSS与最近基因TSS之间的中点,但单向不超过最大延伸距离。 adv_oneDistance: 最大延伸距离(单位:kb)
🧠 使用建议
方法 | 优点 | 缺点 | 推荐使用场景 |
|---|---|---|---|
Basal plus extension | 能捕获远端调控关系,灵活性高 | 可能导致一个 peak 关联多个基因 | 默认首选,特别是做增强子分析时 |
Two nearest genes | 平衡了扩展性和特异性 | 对密集区域仍可能有歧义 | 基因密集区、初步分析 |
Single nearest gene | 简单直观,结果明确 | 忽略远端调控,信息丢失严重 | 快速验证、教学演示 |
选择哪种策略取决于你的实验目的:
- 如果你想探索远端增强子的作用 → 选 Basal plus extension
- 如果你希望减少假阳性 → 选 Two nearest genes
- 如果你只需要快速定位最近基因 → 选 Single nearest gene
运行代码示例
# 读取BED文件并转换为GRanges对象
bed_file <- "HCSE.bed"
peaks_gr <- rtracklayer::import(bed_file, format = "BED")
# 检查读取结果
print(peaks_gr)
#################
#调控区域进行基因注释
###############################
#三种不同模式
job = submitGreatJob(peaks_gr, species = "hg38",adv_upstream = 500, adv_downstream = 500, adv_span = 1500)### 如果是普通增强子,可以选择上下游100kb
job = submitGreatJob(bed, species = "hg38",rule = "twoClosest", adv_twoDistance = 2000)
job = submitGreatJob(bed, species = "hg38",rule = "oneClosest", adv_oneDistance = 2000)
#分析每一个区域注释基因数量的分布
res = plotRegionGeneAssociationGraphs(job)
#将注释结果转换成数据框格式
#R4
.1.5版本下下载的rGREAT,注释基因获取代码
SEHC_gene<-as.data.frame(res)
#R4
.5.2版本下rGREAT包,注释基因获取代码
SEHC_gene <- job@association_tables$all
#下载结果
write.csv(SEHC_gene, "SEHC_gene.csv", row.names = FALSE)
########
#GO富集分析
#########################
tb = getEnrichmentTables(job, download_by = "tsv")
HC_SE_BP<-as.data.frame(tb[["GO Biological Process"]])
write.csv(HC_SE_BP, "HC_SE_BP.csv", row.names = FALSE)基因注释结果
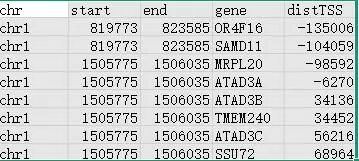
1.peak位点注释基因文件:表格中有超级增强子区域信息,注释的基因名称与超级增强子到基因TSS的距离。
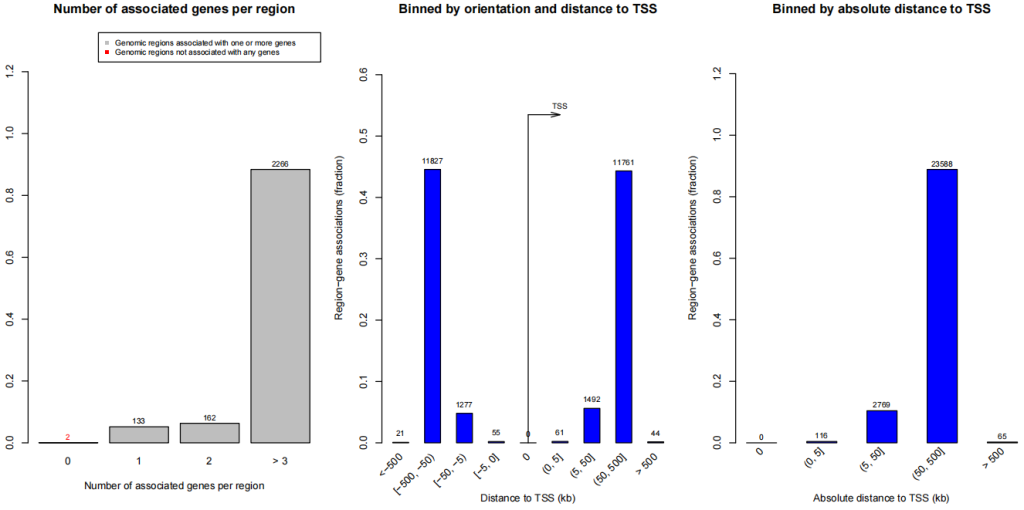
2.在进行plotRegionGeneAssociationGraphs 时候,会绘制peak与基因关联比例关系。如一个peaks 关联多少个基因。调控区域距离基因TSS的距离等。
3.GO富集文件列名解释:
Ontology | 本体类别 | 指明该条目属于哪个本体库,如 GO Biological Process 、GO Molecular Function 或 GO Cellular Component |
|---|---|---|
ID | 术语ID | 基因本体数据库中该条目的唯一标识符,例如 GO:0050789。 |
Desc | 描述 | 对该基因集(Gene Set)的生物学功能或过程的简要描述, |
BinomRank | 二项分布检验排名 | 基于二项分布检验的排名,数字越小,排名越高(越显著)。 |
BinomP | 二项分布原始P值 | 二项分布检验的原始P值,表示在随机情况下观察到当前富集程度的概率,数值越接近0越显著(通常P < 0.05)。 |
BinomBonfP | 二项分布Bonferroni校正P值 | 经过Bonferroni校正后的二项分布P值,用于控制多重假设检验的假阳性率,校正较严格。 |
BinomFdrQ | 二项分布FDR校正Q值 | 经过FDR(错误发现率)校正后的二项分布Q值,是高通量分析中常用的校正方法,通常Q < 0.05认为结果可靠。 |
RegionFoldEnrich | 区域富集倍数 | 区域富集倍数,表示实际观察到的区域数相对于期望数量的倍数,数值越大富集越明显。 |
ExpRegions | 期望区域数 | 期望区域数,根据背景基因组大小,统计模型预测应在此基因集中找到的区域数量。 |
ObsRegions | 实际观察区域数 | 实际观察区域数,在输入数据中实际落在该功能分类下的区域数量。 |
GenomeFrac | 基因组占比 | 基因组占比,该基因集在参考基因组(或背景基因集)中所占的比例。 |
SetCov | 基因集覆盖率 | 基因集覆盖率,该基因集在输入数据集中所占的比例。 |
HyperRank | 超几何分布检验排名 | 基于超几何分布检验的排名,数字越小,排名越高(越显著)。 |
HyperP | 超几何分布原始P值 | 超几何分布检验的原始P值,判断富集是否具有统计学意义。 |
HyperBonfP | 超几何分布Bonferroni校正P值 | 经过Bonferroni校正后的超几何分布P值,控制多重检验的假阳性。 |
HyperFdrQ | 超几何分布FDR校正Q值 | 经过FDR校正后的超几何分布Q值,衡量结果在多重检验中的可靠性。 |
GeneFoldEnrich | 基因富集倍数 | 基因富集倍数,表示实际观察到的基因数相对于期望数量的倍数,反映富集强度。 |
ExpGenes | 期望基因数 | 期望基因数,根据背景基因组预测应在此基因集中找到的基因数量。 |
ObsGenes | 实际观察基因数 | 实际观察基因数,在输入数据中实际落在该功能分类下的基因数量。 |
TotalGenes | 总基因数 | 总基因数,可能指该基因集在背景基因组中的总基因数量。 |
GeneSetCov | 基因集覆盖率 | 基因集覆盖率,该基因集在输入数据集中所占的比例(与SetCov类似,可能针对基因层面)。 |
TermCov | 条目覆盖率 | 条目覆盖率,通常指该GO术语覆盖的基因数量或比例,反映该功能的普遍性。 |
📝 一句话总结
针对传统基于基因的富集分析因假设基因选取概率均等而产生偏差的问题,GREAT算法通过将基因转化为基因组区间并基于二项分布进行检验,从而直接在基因组区间层面进行更准确的富集分析。
📚 详细总结
传统的基因富集分析方法与 GREAT 算法在原理和假设上的核心区别:
1. 传统富集分析方法
- 操作流程首先根据线性距离,将基因组区间(Genomic Regions)注释到具体的基因上,然后利用超几何分布进行富集检验。
- 核心假设该方法的前提是假设每个基因是独立的,且被选中的概率相同。
- 存在的问题这一假设在现实中往往不成立。由于基因在基因组上的分布和长度不同,导致基因被选中的概率并不均等: 位置效应:如果基因组区间集中在某些区域,远离这些区间的基因被选中的概率极低;反之,附近的基因概率更高。长度效应:较长的基因由于“体积”大,比短基因更容易被基因组区间“击中”并注释到。
2. GREAT 算法
- 解决思路为了克服上述偏差,GREAT 不再通过注释基因间接分析,而是直接在基因组区间层面进行考虑。
- 核心假设其前提假设转变为基因组区间在基因组上是均匀分布的。
- 操作流程 方向转换:将基因转换为特定的基因组区间(即定义每个基因的调控域)。统计检验:基于这种转换,使用二项分布来计算富集显著性。
- 优势:这种方法避免了因基因长度差异和位置聚集导致的偏差,能够更真实地反映基因组区间的生物学功能富集情况。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-12-12,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号