
基于gff特性的Biopython解析提取CDS
你好,我正在尝试从一个fasta文件中提取编码序列,它使用一个gff文件,借助biopython (https://biopython.org/wiki/GFF_Parsing)。
我试过做本教程所描述的事情,但有些事情我似乎因为某些原因而不正确:当我迭代序列记录的特性时,只有'gff_type':'gene‘是被识别的。
下面是我的gff文件的一个示例:
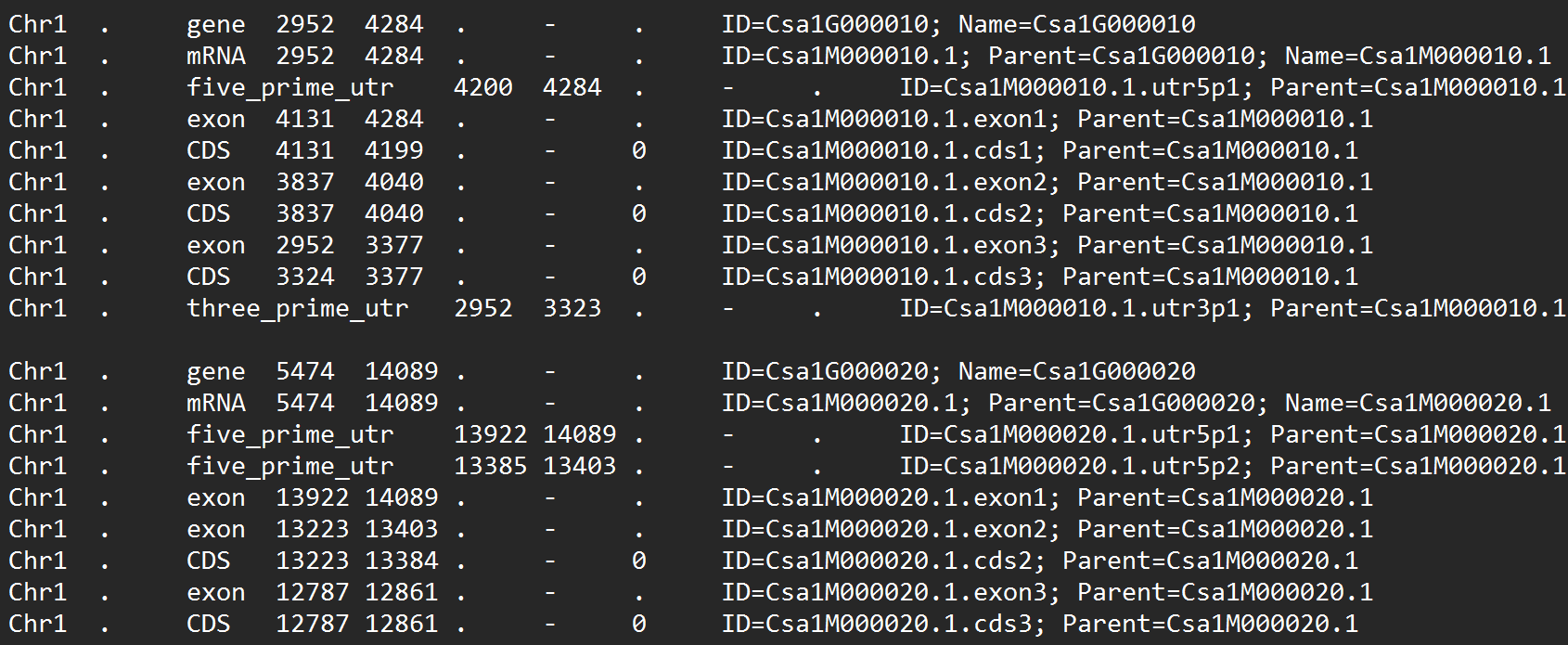
如您所见,我的文件清楚地包含了gff_type='CDS‘条目
但是当我运行这样一个简单的脚本时:
for rec in GFF.parse(in_handle):
for feature in rec.features:
if feature.type == 'CDS':
print(feature)不返回任何输出。当我运行这样的东西时:
for rec in GFF.parse(in_handle):
print(rec.features)
break
SeqFeature(FeatureLocation(ExactPosition(2951), ExactPosition(4284), strand=-1), type='gene', id='Csa1G000010'),
SeqFeature(FeatureLocation(ExactPosition(5473), ExactPosition(14089), strand=-1), type='gene', id='Csa1G000020'),
SeqFeature(FeatureLocation(ExactPosition(18683), ExactPosition(21806), strand=1), type='gene', id='Csa1G000030'),
...有两件事我不明白:
- 为什么只有type=' gene‘特征在
- 上迭代--在我的脚本中有一个中断,所以应该只迭代一个序列记录,如果一个序列记录被确定为所有单个ID的出现(例如Chr1),并且“功能”是这个ID的每个基因,而不是CDS?(CDS和gene都列在可能的gff_type值中,所以我不明白为什么其中一个会比另一个更好)
gff_type: {('CDS',):117359, ('exon',):120675, ('five_prime_utr',):16038, ('gene',):24274, ('mRNA',):24274, ('three_prime_utr',):15588}我觉得我的解决方案是在每个基因中迭代,然后那样提取CDS,但是没有SeqRecord.feature.feature属性。
然而,有一个SeqRecord.feature.sub_feature属性,导致在基因中对mRNA进行迭代(其中只有1),但是没有sub_sub_feature (我用dir检查过)。
所以我被这个问题困住了,这个问题看起来非常简单。我知道我可以像txt一样迭代gff文件,使用选项卡分离,但是我试图更多地了解Biopython,但是没有用(不幸的是,Biopython文档也很糟糕)。我希望有人知道怎么让我上路。
亲切的问候
回答 1
Stack Overflow用户
发布于 2022-06-02 15:08:43
其结构为基因->、mRNA、->外显子、CDS、CDS等3'UTR等序列。外显子或CDS为sub_features of mRNA,而非基因sub_sub_features。
请参阅查看您的GFF文件在这里:https://biopython.org/wiki/GFF_Parsing
https://stackoverflow.com/questions/72263447
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号