Advanced Science | 靶点感知分子生成,到底有没有读懂靶点

Advanced Science | 靶点感知分子生成,到底有没有读懂靶点

MindDance
发布于 2026-05-09 13:48:54
发布于 2026-05-09 13:48:54
最近,浙江大学药学院侯廷军、康玉、王極可等老师在 Advanced Science 发表了题为 Revisiting Target-Aware de novo Molecular Generation with TarPass: Between Rational Design and Texas Sharpshooter 的研究论文。
这篇文章关注的是 AI 药物设计中一个非常现实的问题:当我们把蛋白靶点信息交给生成模型,模型生成的新分子究竟是被靶点真正约束出来的,还是先生成一大批分子,再从里面挑出几个看起来合理的例子进行事后解释?
作者把这种风险写进了标题,借用了 Texas Sharpshooter fallacy,也就是常说的 先射箭后画靶。在 target-aware de novo molecular generation 场景中,它对应一种很容易发生的误判:模型生成了大量分子,研究者再挑出少数 docking score 好、相互作用图漂亮的 case,于是看起来模型似乎学会了靶点信息。但如果这些分子并不比从 ChEMBL 这样的药物样分子库中随机抽样更好,那么所谓的靶点感知就需要被重新审视。

一分钟速读
这篇文章的核心,不是提出一个新的分子生成模型,而是提出了一个更严格的评估框架 TarPass,用来检验 target-aware 分子生成模型到底有没有真正利用靶点信息。
TarPass 不只看 docking score,而是把评估拆成两条线:一条线看生成分子是否能恢复关键 protein-ligand interactions,PLIs;另一条线看分子本身是否合理、类药、可合成,并且是否落在与靶点相关的化学空间里。基于这个框架,作者系统评估了 15 个代表性模型/方法,覆盖 non-3D、3D in situ 和 optimization-based 三类范式。
结论很清醒:3D in situ 模型在预测的蛋白-配体相互作用上平均略有优势,但不少模型并不能稳定优于随机 ChEMBL baseline;non-3D 模型通常更能生成药物样、可合成的分子,但靶点特异性较弱;优化方法可以把分子推向某个性质更优的区域,却常常牺牲其他性质。
换句话说,当前 target-aware 分子生成模型还不能被简单等同于真正的理性设计工具。它们有价值,但更适合作为候选分子来源,再配合严格虚拟筛选、药化判断和实验验证,而不是直接充当自动 hit/lead 发现机器。
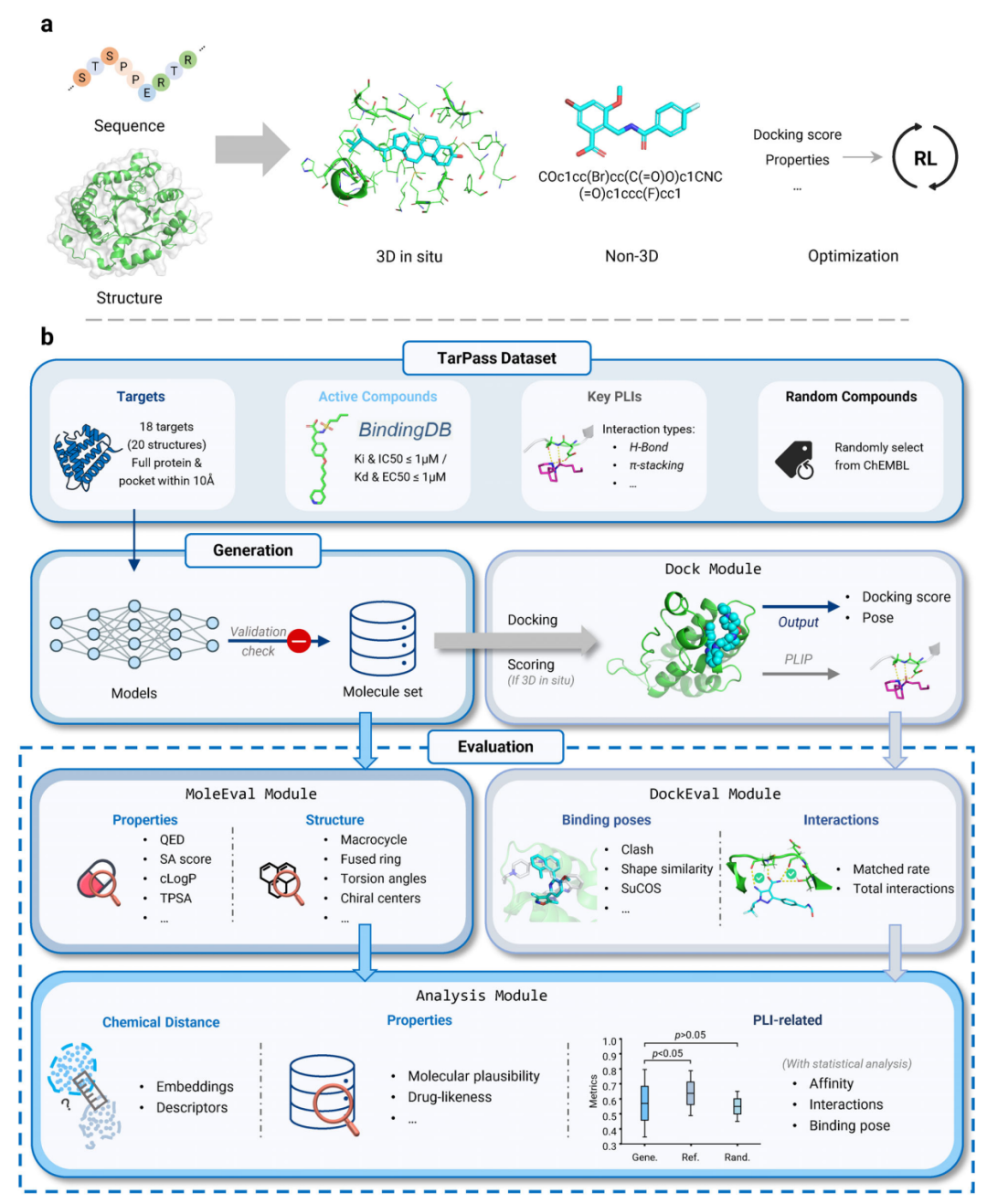
上图左侧概括了 target-aware de novo molecular generation 的三类常见范式:直接在三维口袋中生成配体的 3D in situ 方法、不显式依赖三维几何的 non-3D 方法,以及在生成模型基础上加入后验目标的 optimization-based 方法。下方展示 TarPass 从靶点数据集、分子生成、统一 docking,到 MoleEval、DockEval 和综合分析的完整评估链条
上图左侧概括了 target-aware de novo molecular generation 的三类常见范式:直接在三维口袋中生成配体的 3D in situ 方法、不显式依赖三维几何的 non-3D 方法,以及在生成模型基础上加入后验目标的 optimization-based 方法。下方展示 TarPass 从靶点数据集、分子生成、统一 docking,到 MoleEval、DockEval 和综合分析的完整评估链条
如上图所示,TarPass 的重点不是让某个模型在单一指标上胜出,而是把生成分子放回真实药物发现更关心的多维约束中审视:能不能结合、结合得是否合理、分子本身能不能像一个真实候选物。
为什么
过去几年,生成式模型在药物设计中发展很快。从 SMILES 语言模型、图生成模型,到以蛋白口袋为条件的 3D 扩散模型,研究者希望模型不只是生成看起来像药物的小分子,而是能根据某个具体靶点生成更可能结合的分子。这个方向通常被称为 target-aware molecular generation,如果进一步强调全新设计,也常写作 target-aware de novo molecular generation。
这个方向之所以吸引人,是因为传统早期药物发现往往需要在巨大化学空间中寻找少数可能命中的分子。虚拟筛选可以从已知化合物库中挑分子,但它仍然受限于库里已有的结构。de novo 生成模型的愿景更激进:不只是从库里挑,而是直接提出新分子。DiffSBDD 等结构基础生成模型把 SBDD 表述为三维条件生成问题,用蛋白口袋引导配体生成;TamGen 采用靶点感知的化学语言模型,把蛋白信息和分子序列生成结合起来;早期 DDR1 kinase inhibitor 的生成式设计工作,也让 de novo 分子设计在药物发现领域获得了很高关注度。
但问题在于,会生成分子和会根据靶点设计分子不是一回事。一个模型可以学到药物样分子的统计分布,可以生成高 QED、低 SA score 的结构,也可以在大规模采样后找到 docking score 不错的分子。但这并不自动说明它理解了结合口袋、关键残基、相互作用模式和构象约束。
这里有三个常见误区。
第一,docking score 不是实验活性。Docking 是早期筛选中很有用的计算代理,但它受到打分函数、配体大小、构象搜索、蛋白处理方式等因素影响。文章也明确提醒,TarPass 中的 docking score 和 interaction metrics 应该被理解为统一计算流程下的 proxy,而不是实验亲和力。
第二,漂亮 case 很容易被 cherry-pick。生成模型可以一次生成成千上万个分子,只展示几个成功例子并不能证明整体分布有效。真正有说服力的评估需要把模型输出作为一个集合来分析,并且与参考活性分子和合理随机 baseline 比较。
第三,分子本身合理不代表它对靶点合理。non-3D 模型可能因为预训练充分而生成非常 drug-like 的分子,但缺乏结构约束时,能否形成目标口袋的关键相互作用仍然是问题。反过来,3D in situ 模型可能在口袋里放出一个看似合理的配体,但如果分子包含不合理环系、过多手性中心、空间冲突或不可合成片段,也很难进入真实项目流程。
这正是 TarPass 的切入点:不要只问模型能不能生成分子,而要问它生成的分子是否同时满足靶点相互作用和药物化学合理性。
核心思想:把靶点感知拆成两个可检验问题
文章首先把 target-aware 分子生成形式化为两个互补视角。
训练阶段,模型理想上是在学习蛋白-配体对的联合分布。由此引出第一个猜想:真正 target-aware 的模型应该能在生成分子中编码有意义的 PLIs。这里的 PLI 不只是 docking score 这种隐含亲和力指标,还包括氢键、疏水相互作用、π-stacking、盐桥、卤键等具体物理化学接触。
推理阶段,模型是在给定靶点条件下,从化学空间中采样分子。由此引出第二个猜想:真正 target-aware 的模型不应该只是漫无目的地产生多样分子,而应把输出限制在与该靶点相关、具有生物学意义的化学子空间内。
这两个猜想对应药物设计中的两条经典路线。第一条更接近 structure-based drug design,关注结合口袋、关键残基和相互作用;第二条更接近 ligand-based drug design,关注已知活性分子所在的化学空间和分子性质。TarPass 的设计就是围绕这两条线展开。
先看数据集:18 个靶点,20 个结构,不追求生僻,但追求真实
TarPass 的数据集包含 18 个 well-studied targets,对应 20 个蛋白结构。这些靶点覆盖了药物发现中常见且重要的蛋白类别,包括 7 个激酶、4 个非激酶类酶、3 个 GPCR、2 个核受体和 2 个其他蛋白。作者还强调,这些靶点都与已获批药物或临床候选药物相关,因此具有明确的生物医学相关性。
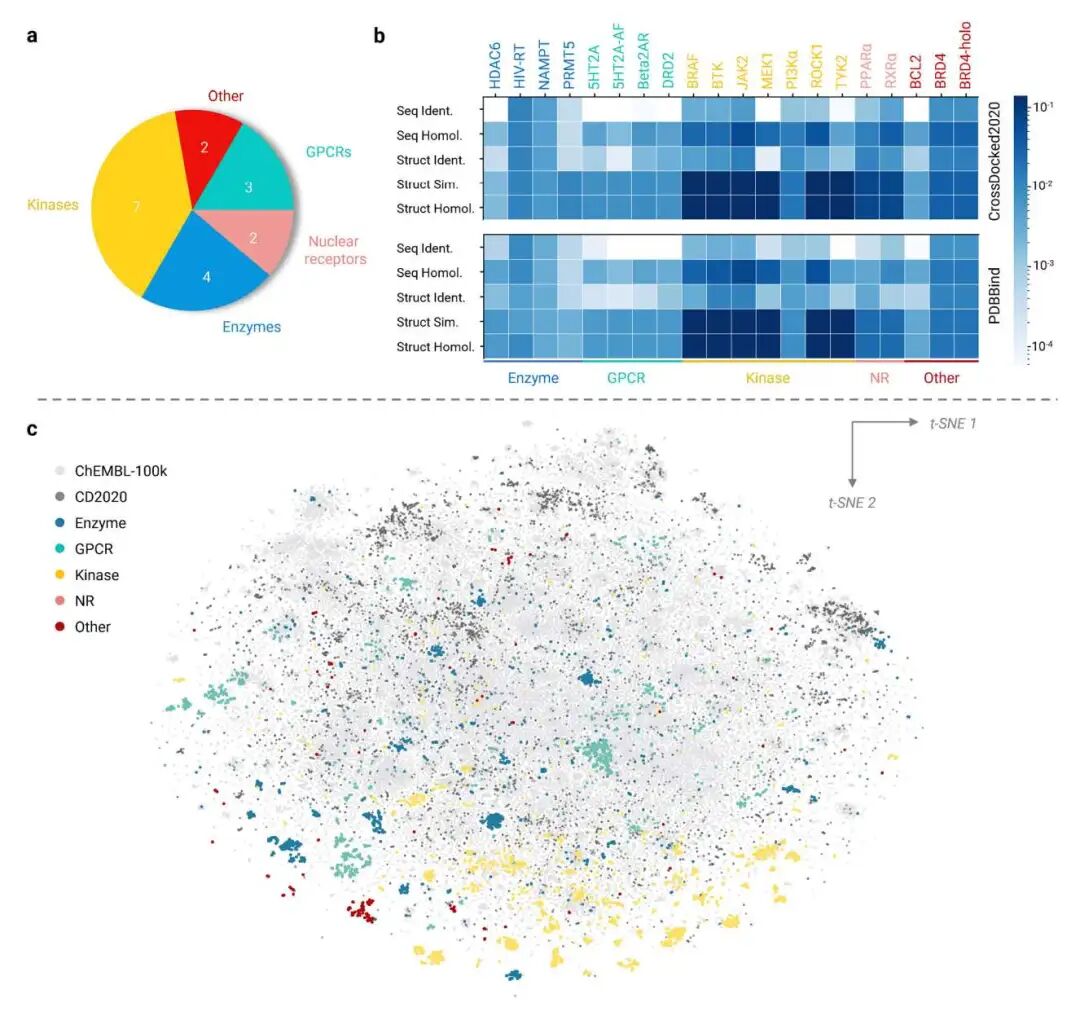
上图展示了 TarPass 测试靶点的蛋白家族分布、测试靶点与 CrossDocked2020/PDBbind 等常用训练集之间的序列和结构相似性,以及 reference actives 在 ChEMBL 背景化学空间中的分布。这个图可以帮助读者理解 TarPass 为什么不是一个只看单个 case 的评估,而是一个尽量贴近真实药物发现约束的 benchmark。
上图展示了 TarPass 测试靶点的蛋白家族分布、测试靶点与 CrossDocked2020/PDBbind 等常用训练集之间的序列和结构相似性,以及 reference actives 在 ChEMBL 背景化学空间中的分布。这个图可以帮助读者理解 TarPass 为什么不是一个只看单个 case 的评估,而是一个尽量贴近真实药物发现约束的 benchmark。
如上图所示,TarPass 并不刻意追求完全陌生、公共数据中几乎没有相似结构的蛋白家族。作者认为,真实药物发现中的可成药靶点往往属于公共数据中已经比较丰富的蛋白家族,例如激酶、GPCR 或核受体。因此,TarPass 更关心的是模型能否在这些现实中高度相关的靶点家族内泛化,而不是在一个人为构造的极端陌生场景中表现。
为了减少数据泄漏,作者从 POKMOL-3D 等资源出发,选取释放时间不早于 2019 年 12 月的结构,并用 MMseqs2 和 Foldseek 对测试靶点与 CrossDocked2020、PDBbind 这类常用训练集做序列和结构相似性分析。结果显示,测试靶点与这些训练集在序列层面的直接重叠很低;在结构层面,激酶等家族存在更高相似性,这正是作者希望保留的现实问题。
TarPass 还加入了两个更贴近真实项目的设置。第一类是 无配体结构与 holo 结构的对照:5-HT2A 使用 AlphaFold 预测结构作为无配体结构,BRD4 使用实验 apo 结构,用来观察模型在缺少共结晶配体参照时的稳定性。第二类是 allosteric targets,包括 MEK1 和 TYK2,用来测试模型面对非经典结合位点时是否仍能生成合理分子。
每个靶点还配套了两类重要信息:一类是从 BindingDB 收集并经过筛选的实验验证活性分子,另一类是结合文献、晶体结构、突变实验和 PLIP 识别结果整理出的关键相互作用。除此之外,TarPass 还加入了从 ChEMBL 随机抽取的 1000 个分子作为 baseline。
TarPass 数据集和对照集合的关键信息
TarPass 数据集和对照集合的关键信息
ChEMBL baseline 尤其重要。文章报告,这 1000 个随机分子的重原子数范围为 4 到 54,平均 28.40;QED 范围为 0.04 到 0.95,平均 0.56,并且其中有 2 个分子与某些靶点的 reference compounds 重叠。正因为如此,这个 baseline 更像一个实际竞争对手:如果生成模型不能比从 ChEMBL 随机抽样更好,它的额外计算成本和复杂建模就需要重新评估。
评估流程:不只看分数,还看姿势、相互作用和分子本身
上面的流程图已经展示了 TarPass 的整体工作流。简单说,每个模型对每个靶点最多进行两轮生成,目标是得到 1000 个 unique molecules。随后,这些分子进入统一 docking 流程。对于 3D in situ 模型,作者还额外评估模型给出的初始构象,因为这类模型本身声称能在口袋中生成三维配体。
TarPass 的评价模块分为两部分。
DockEval 负责 protein-ligand interaction 相关分析,包括 docking score、关键相互作用匹配、结合姿势相似性、空间冲突、中心位置偏移等。作者使用 Gnina 进行 docking,使用 PLIP 检测相互作用,并将检测到的相互作用与人工整理的关键 PLI 列表进行比较。
MoleEval 负责分子本身的合理性分析,包括 QED、SA score、Lipinski rule、PAINS/SureChEMBL/Glaxo alerts、手性中心、螺环、稠环、环大小、heteroatom ratio、torsion angle、化学空间距离等。
这个设计的意义在于,TarPass 不允许模型只在一个维度上好看。一个分子 docking score 好,但结构不合理,不算真正有用;一个分子类药性很好,但无法形成靶点关键相互作用,也不能证明模型懂靶点。
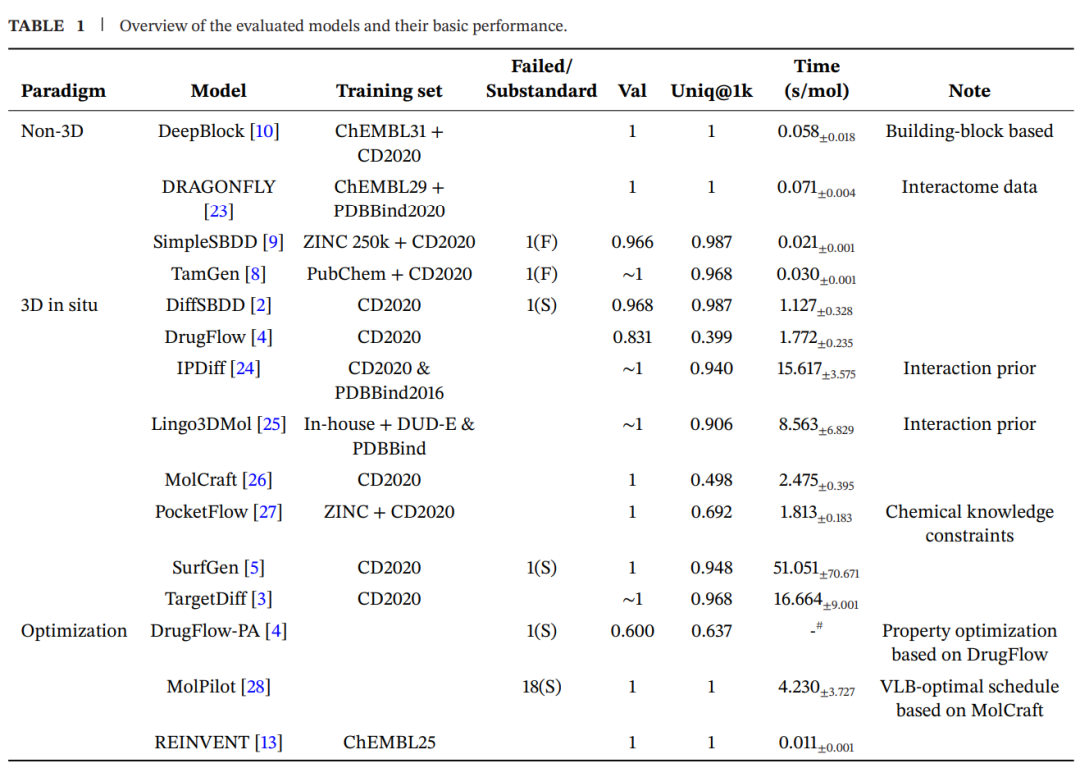
被评估的 15 个模型:三类范式放在同一张考卷上
文章评估了 15 个代表性模型/方法,覆盖三类主流范式。下表按实际评估条目整理,其中 optimization-based 类别包括 DrugFlow-PA、MolPilot 和 REINVENT;DrugFlow-PA 和 MolPilot 分别是 DrugFlow 和 MolCraft 的优化变体。
TarPass 评估的模型范式和代表方法
TarPass 评估的模型范式和代表方法
结果一:生成速度和可部署性差异很大
在真实项目中,模型是否能跑、能不能接受指定结构、能不能稳定生成足够数量的有效分子,本身就是第一道门槛。
TarPass 的基本性能测试显示,non-3D 模型通常速度更快。TamGen 这类模型生成单个分子通常小于 0.1 秒,SimpleSBDD 在作者测试环境中约为 0.021 秒每分子。相比之下,3D in situ 模型明显更慢。DrugFlow 和 PocketFlow 在作者测试环境中约为 1–2 秒每分子,MolCraft 约为 2.5 秒每分子;IPDiff 和 TargetDiff 超过 10 秒;SurfGen 平均达到约 51 秒每分子,并且波动很大。
结构兼容性方面也出现了实际问题。SimpleSBDD 因为预定义 atom vocabulary 中没有金属原子,无法处理 HDAC6 中的锌离子及其他金属环境。TamGen 不接受用户自定义结构,因此不能直接使用 AlphaFold 预测的 5-HT2A 结构作为输入。
分子集合层面,大多数模型能保持较高 validity,但 DrugFlow 系列有效率偏低;non-3D 方法通常能生成高度 unique 的集合,而 DrugFlow、MolCraft、PocketFlow 等 flow-based 模型 uniqueness 较低,提示其分子图采样可能存在重复访问高概率区域的问题。
这部分结果看似基础,却很接近真实部署:模型文章中的性能很重要,但能否处理金属、apo 结构、预测结构,以及能否在固定预算内生成足够多的有效分子,同样决定了它能否进入项目流程。
结果二:3D in situ 平均更会对接,但很多模型仍打不过随机 baseline
Docking score 是最常见的 SBDD 代理指标。TarPass 的结果显示,3D in situ 模型在 docking score 上确实有平均优势。reference ligands 的中位 Vina score 为 -9.109,random ChEMBL baseline 为 -8.217。许多 3D in situ 模型在 redocking 后的平均中位 docking score 位于 -7.5 到 -10 之间,优化类方法甚至可以低于 -10。
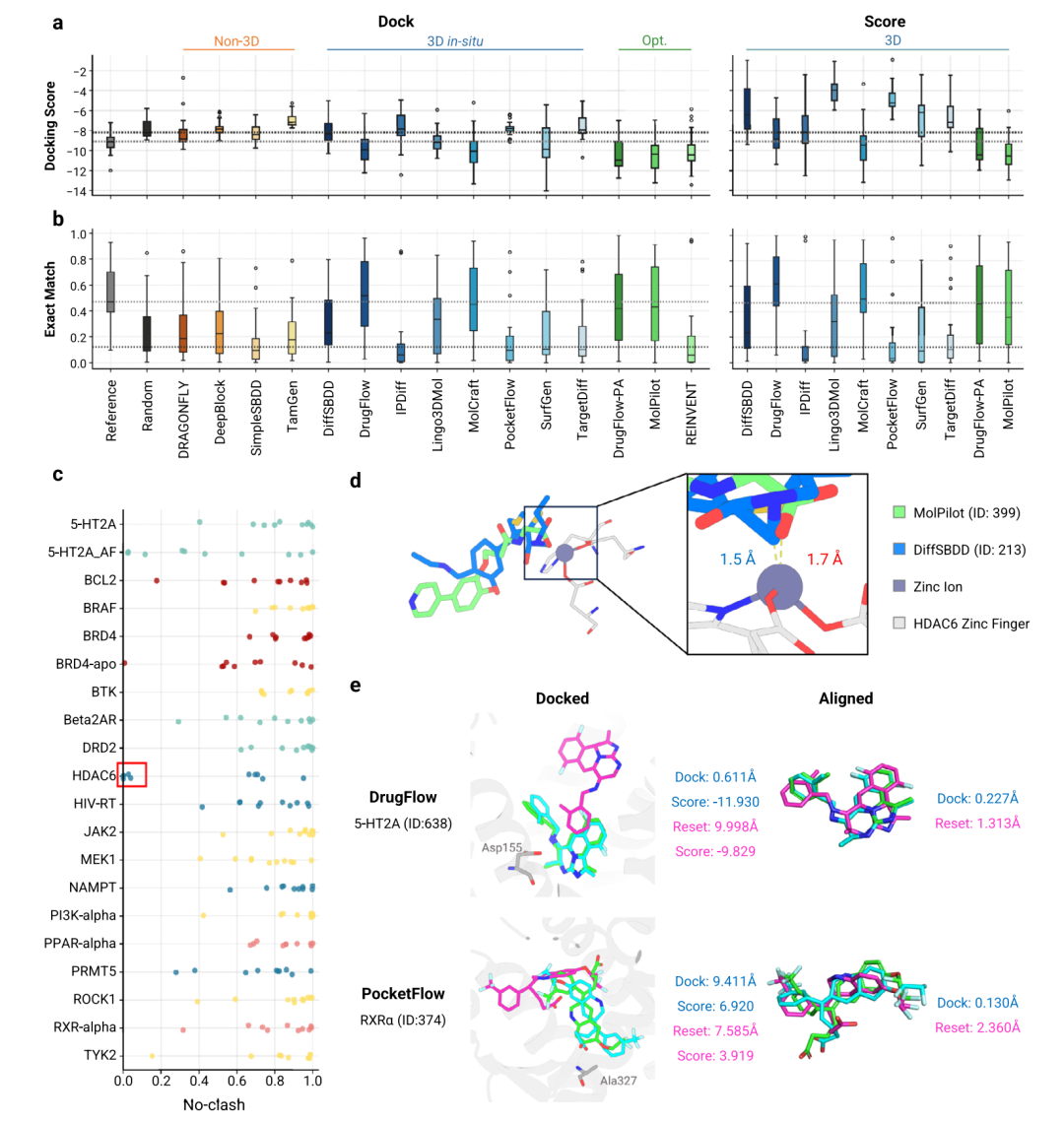
模型在 docking score、关键相互作用恢复和 pose clash 上的表现
模型在 docking score、关键相互作用恢复和 pose clash 上的表现
更关键的是统计比较。作者发现,在超过一半靶点上显著优于随机分子的模型并不多:只有 1 个 non-3D 模型、1 个 optimization-based 模型和 4 个 3D in situ 模型。这说明,即使 3D in situ 在平均上有优势,很多模型也未能稳定超过从 ChEMBL 中随机抽取的药物样分子。
这里不能简单说模型没用。由于 ChEMBL baseline 本身偏向 bioactive chemical space,这个随机对照并不弱。更准确的解读是:当前 target-aware 生成模型如果只用 docking score 证明自己,证据还不够强。
结果三:恢复关键相互作用,比拿到好 docking score 更难
TarPass 更严格的一点,是它不只看 docking score,还看模型是否恢复专家标注的关键 PLIs。
作者使用两个指标描述相互作用恢复能力。Exact Match 衡量所有指定关键相互作用是否被准确复现,要求较严格;Match Ratio 更宽松,衡量任意关键相互作用的恢复比例。
结果比 docking score 更不乐观。由于 docking protocol 和 PLIP 几何阈值本身会带来误差,reference ligands 的 exact match rate 也只有 51.4%;但这仍然是 random baseline 22.3% 的两倍以上。因此,关键相互作用匹配仍然是有意义的评价标准。
在生成模型中,大多数模型的相互作用恢复能力并不优于随机分子。DrugFlow、MolCraft 以及它们的优化变体表现较好,接近 reference ligands 水平。带 interaction prior 的模型表现并不一致:IPDiff 低于 random baseline,而 Lingo3DMol 优于若干 3D in situ 方法。这提示一个很重要的问题:不是往模型里加入 interaction prior 就一定有效,prior 的物理定义和实现方式非常关键。
REINVENT 在相互作用恢复上低于 random baseline,作者认为这可能与其强化学习奖励主要聚焦亲和力有关。也就是说,直接优化 affinity proxy 并不保证关键相互作用正确。
文章还提醒,DrugFlow 和 MolCraft 的强表现可能与原始输入 ligand 高度相关。作者进一步测试后发现,这类表现可能依赖输入 ligand 和大量采样,因此其稳健性和泛化性仍需谨慎看待。
结果四:初始构象和结合姿势仍是 3D 生成模型的痛点
3D in situ 模型最大的卖点,是在蛋白口袋中直接生成三维分子。按理说,它们不只是给出分子图,还应该给出合理 pose。但 TarPass 的 pose-level 分析显示,这仍然是当前模型的薄弱环节。
作者用 PoseBusters 中的标准检查 steric clash。结果显示,大多数 3D in situ 模型生成的初始 pose 都存在一定比例的空间冲突。在基础 3D in situ 模型中,除了 IPDiff,没有方法的 no-clash rate 超过 85%。优化方法能改善这一点,DrugFlow-PA 和 MolPilot 分别相较基础模型提高约 13% 和 8%,并且都超过 90%。
一些失败案例非常有代表性。对于 5-HT2A_AF,部分模型使用 10 Å cutoff 截取 pocket,导致忽略了完整结合口袋中的 ECL2 区域,从而出现 clash。对于 HDAC6,问题集中在 zinc finger domain:多数模型要么忽略锌离子,要么无法正确捕捉 zinc-ligand coordination。上图中展示的 HDAC6 案例里,DiffSBDD 生成的碳原子距离锌离子 1.5 Å,MolPilot 生成的氧原子距离锌离子 1.7 Å;后者短于晶体结构中常见的大于 2 Å 的 Zn-O 键长,前者也表现为不合理的近距离接触,反映出模型对金属配位环境处理不足。
结合模式相似性方面,DrugFlow 和 MolCraft 的表现接近 reference ligands,而 non-3D 模型以及 IPDiff、PocketFlow 等部分 3D 模型接近 random baseline。作者进一步发现,redocked pose 的 centroid displacement 与 Exact Match 之间存在很强负相关,R² = 0.836。这说明分子中心偏得越远,越难维持关键残基相互作用。
作者还设计了 conformation-reset docking:先丢弃模型初始构象,用 MMFF94 重新生成构象后再 docking。结果显示,所有模型在这种流程下的 affinity、interaction match 和 binding mode similarity 都下降,但相对排名大体保持。这说明初始 pose 对结果有影响,而 reset docking 可以作为一种补充评估,帮助区分模型是否真正学到了目标口袋约束,而不是受初始构象偏置影响。
结果五:non-3D 更像药,3D 模型更容易生成奇怪结构
如果说 PLI 是模型是否懂靶点,那么 molecular plausibility 就是在问:模型生成的分子能不能像一个真实药物发现项目中的候选物。
TarPass 在 2D 结构层面做了很细的检查,包括手性中心、螺环、稠环、环大小、柔性、heteroatom ratio 等。整体趋势很清楚:预训练充分的模型,尤其是 non-3D 模型,更容易生成接近真实药物样化学空间的分子。
真实分子通常平均少于 1 个手性中心,spiro atoms 也很少。non-3D 模型大体复现了这一趋势,而一些 3D in situ diffusion-based 模型生成了过多手性中心和 spiro atoms,明显削弱分子合理性。环系统也类似。多数模型平均生成 3–5 个环,接近真实分子;但 acyclic molecules 在 reference ligands 中几乎没有,生成模型中却明显更多。DiffSBDD 生成 undesired rings 的比例达到 42%,SurfGen 中含高度稠环系统的分子比例达到 74%,这会显著增加合成和实际存在的风险。
药物样性方面,non-3D 模型的 QED 通常更高,多数超过 0.6;reference compounds 的平均 QED 为 0.474。Lipinski rule 方面,大多数模型接近真实分子,约 3.9 条通过;但 DrugFlow-PA 和 REINVENT 是例外。SA score 的差异更明显:序列类模型更接近 reference 平均值 3 左右,而多数 graph-based 3D in situ 模型超过 3.5,SurfGen 和 DiffSBDD 超过 4。DrugFlow-PA 通过性质优化使 SA score 相比基础模型降低约 0.9,也把 SureChEMBL alerts 相比基础模型降低 3 倍以上。
这个结果并不是说 non-3D 一定比 3D 好,而是说明 3D 口袋约束并不能自动带来药化合理性。如果训练数据覆盖不足、化学先验不够强,3D 模型很容易在空间上生成看似可行但药化上不舒服的分子。
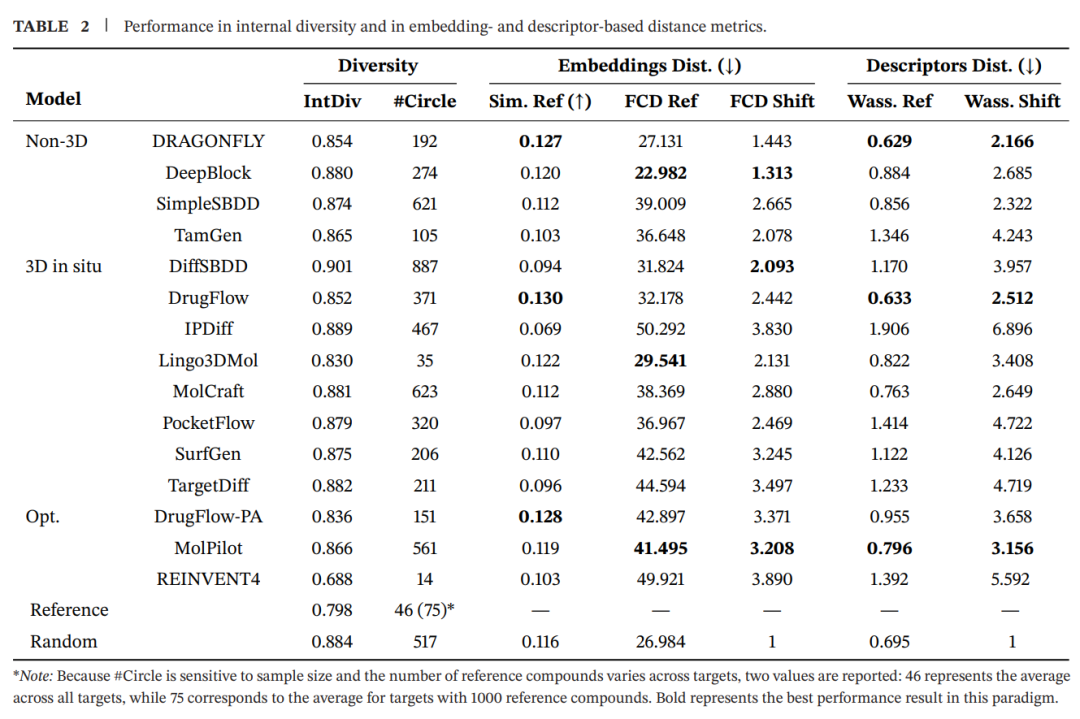
结果六:化学空间分析提示,多样性不是越大越好
生成模型常常强调 novelty 和 diversity。但在靶点感知药物设计中,多样性必须有边界。分子可以新,但不能偏离已知 active compounds 和 drug-like chemical space 太远。
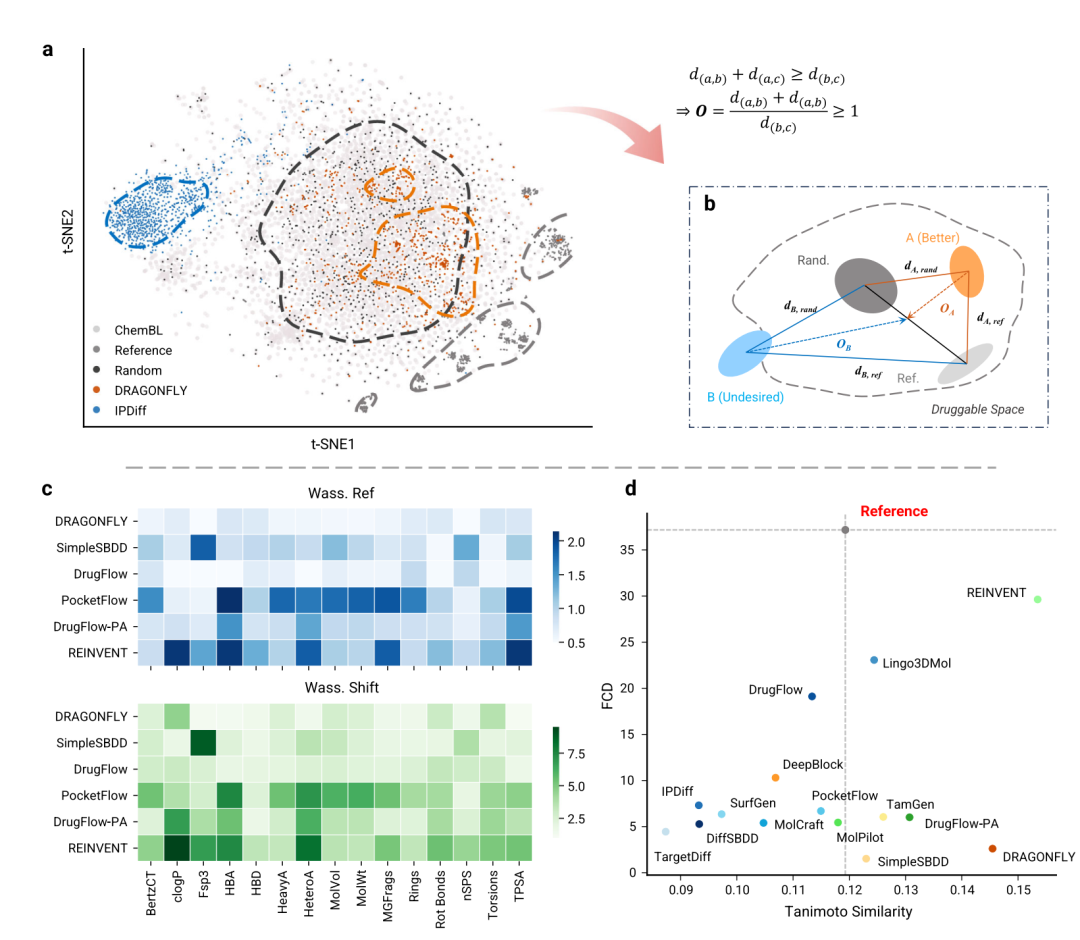
化学距离、shift index 与跨靶点相似性分析
化学距离、shift index 与跨靶点相似性分析
TarPass 用多个化学距离指标评估这一点,包括 ECFP4 Tanimoto similarity、Fréchet ChemNet Distance,FCD,以及作者引入的基于 15 个描述符的 Wasserstein distance。作者还设计了一个 shift index,用来衡量生成分布相对 reference actives 和 random ChEMBL baseline 的偏移程度。
结果显示,IntDiv 对模型差异不太敏感,许多模型位于 0.85–0.9,与 random molecules 接近,高于 reference actives 的 0.798。相比之下,#Circle 更能反映问题。对于拥有 1000 个 reference compounds 的靶点,reference #Circle 平均约 75,而 random molecules 为 517。多数模型超过 100,DiffSBDD 和 MolCraft 甚至超过 random baseline,提示这些模型生成的分子集合可能过于分散。
FCD 和 Wasserstein distance 给出了更可解释的结果。部分 ChEMBL 预训练的 non-3D 模型,例如 DeepBlock 和 DRAGONFLY,距离真实分子更近,shift 值较低;容易生成不理想结构的模型 shift 更高。优化类方法也可能出现更高 shift,说明单一性质优化会把分子推离 drug-like chemical space。例如 REINVENT 在 clogP 上偏移明显,DrugFlow-PA 在氢键受体、heteroatom count 和 TPSA 上偏移明显。
这部分给出的药化启示很直接:novelty 本身不是目标。只有在保留关键相互作用和基本药物样性质的前提下,结构多样性才有意义。
结果七:靶点特异性有家族层面的信号,但细粒度仍然不足
真正的 target-aware 模型应该能对不同靶点生成不同分子,尤其是在结构相似但结合位点或关键相互作用不同的情况下。
TarPass 首先从蛋白家族层面分析生成分子的相似性。结果显示,在 non-3D 和 3D in situ 两类范式中,kinases 和 GPCRs 的 family 内 FCD 都较低,说明模型在一定程度上能捕捉同一家族靶点的相似结构特征。两因素 ANOVA 也显示,模型和靶点都会影响生成分子的 PLI 表现,并且存在交互作用。这说明 target specificity 并非完全不存在。
但更细粒度的测试暴露了问题。作者构建了两个无配体结构与 holo 结构的对照,以及一个 site-specific 对照:5-HT2A_AF 与 5-HT2A、BRD4-apo 与 BRD4-holo,以及 JAK2-TYK2。前两组结构 RMSD 分别为 0.402 Å 和 0.304 Å,理论上生成分子应保持一致;JAK2 和 TYK2 同属 JAK family,但一个对应 ATP-binding pocket,另一个对应 pseudokinase domain,理论上生成分子应体现特异性。
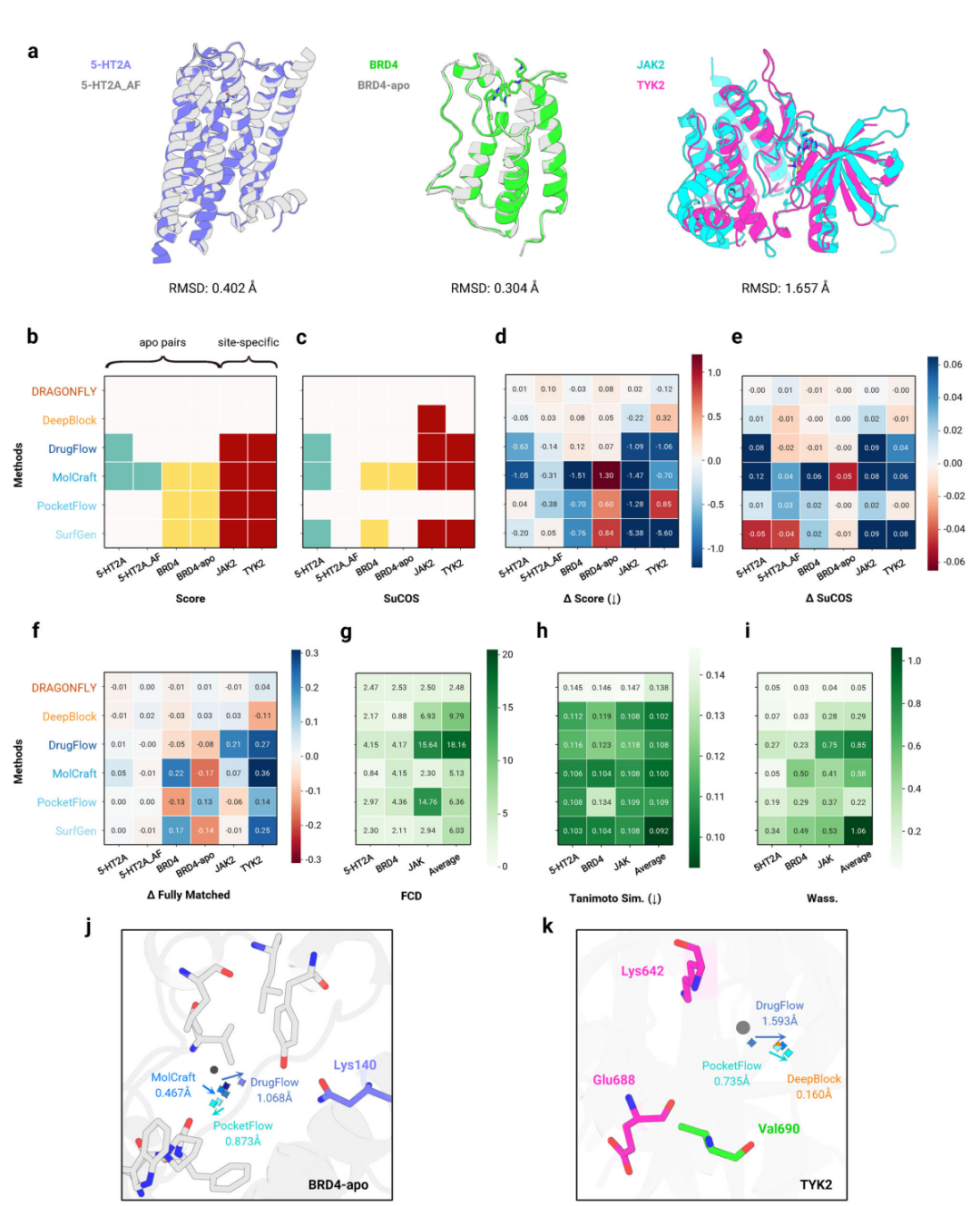
结构相似靶点中的一致性与特异性测试
结构相似靶点中的一致性与特异性测试
在 FCD 分析中,理想情况是无配体结构与 holo 结构对照的 FCD 更低,而 JAK2-TYK2 的 FCD 更高。整体上,只有 DeepBlock、DrugFlow 和 PocketFlow 基本符合这个预期;其中 DrugFlow 还依赖原始 ligand 采样。cross-docking 分析进一步显示,non-3D 模型通常难以产生明显差异;3D in situ 模型在 5-HT2A 上较一致,但在 BRD4 对照和 JAK2-TYK2 上表现差异较大。
上图中有一个细节很值得看:在 BRD4-apo 和 TYK2 的例子中,非原生靶点生成分子的 ligand centroid 会偏离关键残基,导致 PLI 表现下降。这说明 3D in situ 模型可能对局部 pocket 环境过于敏感,还没有稳定地学会在相似结构中识别关键差别。
因此,TarPass 对 target specificity 的结论比较克制:当前模型在蛋白家族层面有一定信号,但是否能实现细粒度、真正靶点特异性的生成,仍需要更系统验证。
多层虚拟筛选:当前模型更适合做候选分子来源
文章并没有停留在批评模型。作者进一步问了一个很实际的问题:既然当前模型还不完美,那它们生成的分子还能不能通过后处理变得有用?
为此,作者设计了一个 multi-tier virtual screening workflow,把模型输出先经过硬过滤,再经过经验性软过滤,最终缩小到更适合实验验证的规模。测试对象是 JAK2 和 TYK2。
第一层硬过滤包含三类条件。PLI 层面要求 docking 和 rescoring scores ≤ -8,并且满足所有预定义关键相互作用;结构层面要求分子完整、只含常见原子、最大环不超过 7 个原子、分子量 250–750;药物样性层面要求 SA score ≤ 4.0、QED ≥ 0.4、至少满足 4 条 Lipinski rules,并且没有 structural alerts。
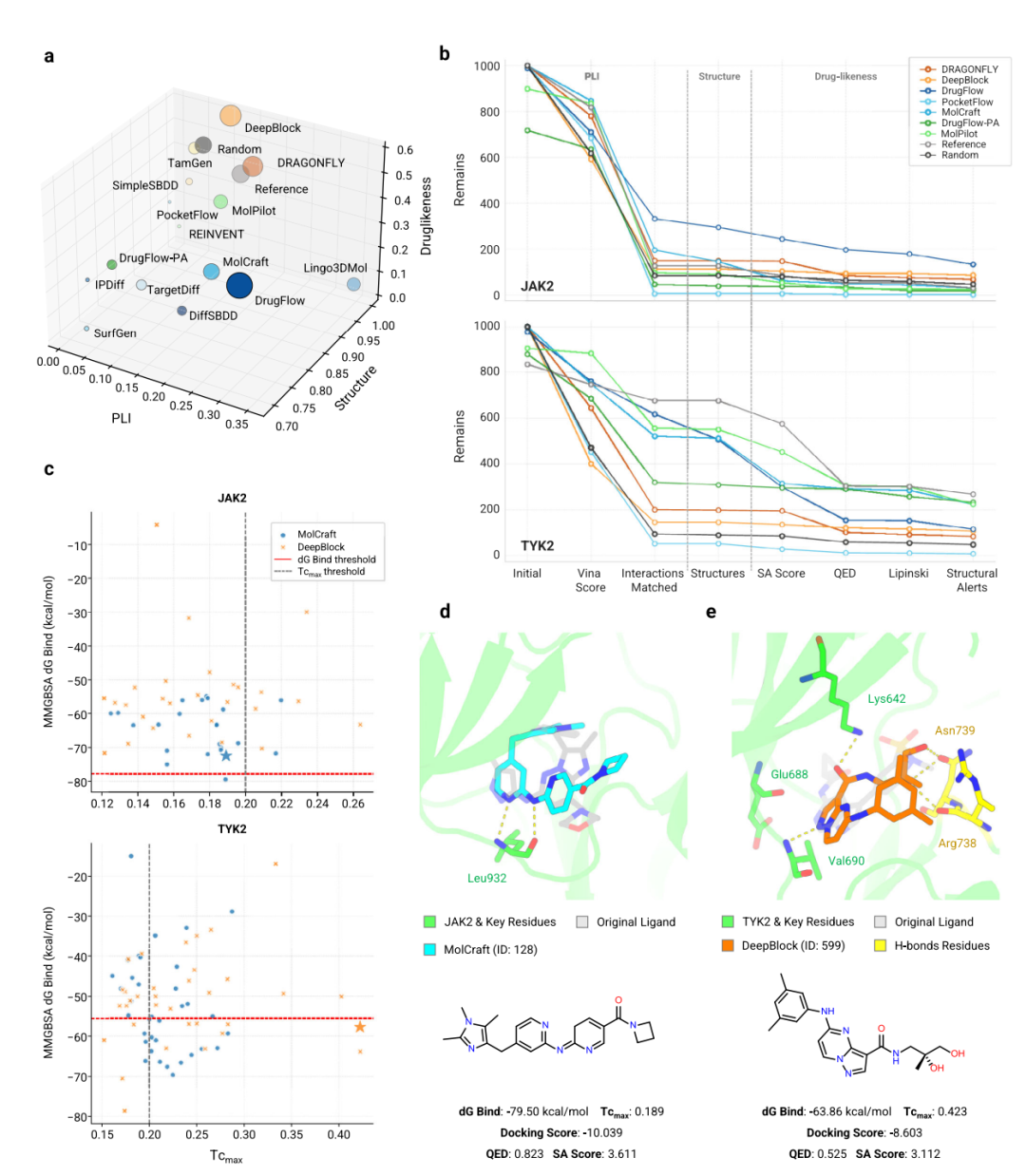
多层虚拟筛选流程及 JAK2/TYK2 代表候选分子
多层虚拟筛选流程及 JAK2/TYK2 代表候选分子
结果显示,在第一层硬过滤后,3D in situ 和 non-3D 范式都能保留约 10% 的分子。这个比例说明两类方法都存在实际使用价值,但剩余候选仍然太多。作者进一步选取表现较好的 MolCraft 和 DeepBlock 做精细筛选。由于这两个模型输出的 internal diversity 很高,单纯 2D clustering 效果不佳,作者转而使用经验性 soft filters,并结合 MM/GBSA binding free energy 和 Tcmax 进行回顾性评估。Tcmax cutoff 设为 0.2,用来平衡新颖性和靶点相关性。
最终,JAK2 和 TYK2 都观察到一定 enrichment,TYK2 的候选中更多分子同时满足 affinity 和 similarity 标准。上图展示的两个代表分子结构上较合理,pose 也能解释一定靶点结合可能性。但作者也明确指出,它们离强 lead 还有距离:JAK2 分子缺乏额外稳定相互作用,TYK2 分子没有充分占据决定选择性的 alanine pocket。
这部分结论很重要:多层虚拟筛选能提高当前生成模型的可用性,但它不是替代模型进步的万能补丁。真正需要提高的仍然是 pose 准确性、相互作用保真度和分子合理性。
启发
第一,target-aware 不等于 target-understanding。只要模型输入了蛋白结构或序列,就把它称为理解靶点,是不严谨的。至少应该检查关键 PLIs、pose 合理性、target-specific chemical space,以及是否超过合理 baseline。
第二,随机 baseline 必不可少,而且要选得现实。TarPass 使用 ChEMBL random baseline 很有意义,因为真实药物发现中,生成模型面对的竞争对手往往不是纯噪音,而是已有 bioactive-like compound space。能不能打过这样的 baseline,决定了生成模型是否真的带来增益。
第三,docking score 不能单独承担证明责任。这篇文章清楚显示,好的 docking score 不保证关键相互作用恢复,也不保证初始 pose 没有 clash,更不保证分子可合成。对于 target-aware 生成模型,评价应该同时覆盖 affinity proxy、interaction pattern、pose plausibility 和 medicinal chemistry properties。
第四,预训练和化学先验非常重要。non-3D 模型在分子合理性上的优势,主要来自更广泛的化学空间预训练。3D in situ 模型如果缺少足够化学先验,很容易生成空间上看似贴合但药化上不舒服的结构。未来模型需要把三维结构约束和大规模药物样分子先验更好地结合。
第五,优化不能只盯单一目标。DrugFlow-PA 和 REINVENT 的结果说明,单一性质优化确实能改善某些指标,但可能让其他性质变差。药物设计天然是多目标问题,亲和力、选择性、可合成性、稳定性、ADMET 风险和 novelty 都需要平衡。
第六,生成模型目前更像候选空间扩展器,而不是自动理性设计器。从这篇文章的结果看,把 target-aware generation 用作 molecule source,再结合虚拟筛选、药化规则、专家判断和实验验证,是更稳妥的路线。
总结
这篇论文最有价值的地方,不是给 15 个模型排出一个简单名次,而是把 target-aware molecular generation 从漂亮案例拉回到了可验证、可比较、可复现的评估框架里。
TarPass 传递的信息可以概括为一句话:真正的靶点感知分子生成,不能只生成看起来像药的分子,也不能只追求 docking score;它必须同时经得起关键相互作用、结合姿势、分子合理性、化学空间和靶点特异性的检验。
对于 AI 药物设计研究者而言,这篇文章是一次很必要的提醒。生成模型当然会继续进步,但在走向真实药物发现之前,我们需要更少的漂亮 case,更多的严格 benchmark;更少的事后解释,更多的可重复评估;更少的单指标优化,更多的多目标、物理化学一致性和药化可行性。
这也正是 TarPass 的意义所在:它没有否定 target-aware 分子生成,而是把这个领域往更接近真实药物发现的方向推了一步。
参考资料
R.Qin, Z.Chen, Y.Li, et al. “Revisiting Target-Aware de novo Molecular Generation with TarPass: Between Rational Design and Texas Sharpshooter.” Advanced Science (2026): e75411. https://doi.org/10.1002/advs.75411
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-29,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号