分析梳理--分子动力学模拟的常规步骤七(Gromacs)
原创
分析梳理--分子动力学模拟的常规步骤七(Gromacs)
原创
追风少年i
发布于 2026-04-30 08:40:26
发布于 2026-04-30 08:40:26
作者,Evil Genius
这一篇我们继续分子动力学。
NPT预平衡密度值与标准结果不同,是因为与压力有关的性质收敛一般很慢,因此运行NPT平衡的时间可以适当延长一些。
经过了前面完整的处理之后,我们就需要对我们的MD的结果进行一定的基础分析了。
简单再回顾一下md.mdp文件(这个参数的设置非常重要)
title = OPLS Lysozyme NPT Production
; Run parameters
integrator = md
nsteps = 50000 ; 100 ps(生产跑通常更长,按需调整)
dt = 0.002
; Output control(关键:必须指定 xtc 输出)
nstxout = 0 ; 不输出未压缩坐标
nstvout = 0
nstfout = 0
nstxout-compressed = 5000 ; ✅ 每 5000 步写一次 .xtc(生成轨迹)
nstenergy = 5000
nstlog = 5000
; Bond parameters
continuation = yes ; ⚠️ 生产跑:从已有速度继续
constraint_algorithm = lincs
constraints = all-bonds
lincs_iter = 1
lincs_order = 4
; Neighbor searching / electrostatics(建议补齐,避免用默认值)
cutoff-scheme = Verlet
ns_type = grid
rlist = 1.0
coulombtype = PME
rcoulomb = 1.0
vdwtype = Cut-off
rvdw = 1.0
; Temperature coupling
tcoupl = V-rescale
tc-grps = Protein Non-Protein
tau_t = 0.1 0.1
ref_t = 300 300
; Pressure coupling(保持不变,说明你确实要跑 NPT)
pcoupl = Parrinello-Rahman
pcoupltype = isotropic
tau_p = 2.0
ref_p = 1.0
compressibility = 4.5e-5
refcoord_scaling = com
; PBC & dispersion
pbc = xyz
DispCorr = EnerPres
; Velocity generation(生产跑:不从零生成速度)
gen_vel = no首先是处理周期性
蛋白质将通过晶胞扩散,并可能出现“破碎”或“跳跃”到盒子的另一侧。要解决此类行为,使用以下命令:
gmx trjconv -s md_0_I.tpr -f md.xtc -o md_0_I_noPBC.xtc -pbc mol -ur compact选择0(“系统”)作为输出。将对这条“修正过的”轨迹进行所有分析。
gmx triconv可以通过多种方式转换轨迹文件,它用作后处理工具,用于去除坐标、校正周期性或手动更改轨迹(时间单位、帧频率等)
首先解读一下选项
组号 | 组名 | 原子数 | 含义与用途 |
|---|---|---|---|
0 | System | 33876 | 整个系统的所有原子。几乎所有操作都可以选这个,但通常不推荐,因为会把水和离子也包含进去。 |
1 | Protein | 1960 | 蛋白质的全部原子。最常用的组之一,用于分析蛋白质本身的结构(如RMSD、回旋半径)。 |
2 | Protein-H | 1001 | 蛋白质中所有氢原子。比较少单独使用,可能用于分析氢键或特定光谱。 |
3 | C-alpha | 129 | 蛋白质的Cα原子。每个残基一个,共129个(暗示你的蛋白质有129个残基)。非常常用,用于计算蛋白质的主链构象变化(RMSD),可消除侧链影响。 |
4 | Backbone | 387 | 蛋白质的主链原子(C、CA、N)。常用于比Cα更细致的RMSD分析。 |
5 | MainChain | 517 | 主链+连接主链的氢?通常和Backbone很接近。 |
6 | MainChain+Cb | 634 | 主链原子加上每个残基的Beta碳(CB)。用于某些需要考虑侧链起始方向的分析。 |
7 | MainChain+H | 646 | 主链原子加上与主链相连的氢原子。 |
8 | SideChain | 1314 | 所有侧链原子(即蛋白质中除主链以外的原子)。用于分析侧链的排布或柔性。 |
9 | SideChain-H | 484 | 侧链上的氢原子。 |
10 | Prot-Masses | 1960 | 与Protein原子数相同,用于质心校正等高级选项。通常不需要直接选择。 |
11 | non-Protein | 31916 | 所有非蛋白质原子(即水+离子)。用于将溶剂和蛋白质分开处理。 |
12 | Water | 31908 | 所有水分子中的原子(包括O和H)。用于分析溶剂行为或仅输出水轨迹。 |
13 | SOL | 31908 | 同Water,SOL是GROMACS内部对水分子的标准命名。 |
14 | non-Water | 1968 | 所有非水原子(蛋白质+离子)。和non-Protein相反。 |
15 | Ion | 8 | 离子原子。这里有8个离子,很可能是为了中和蛋白质电荷而加入的(例如8个Cl⁻或8个Na⁺)。 |
16 | Water_and_ions | 31916 | 水+离子。用于需要将溶剂整体考虑(但排除蛋白质)的分析。 |
然后看一下gmx trjconv 的参数
Options to specify input files:
-f [<.xtc/.trr/...>] (traj.xtc)
Trajectory: xtc trr cpt gro g96 pdb tng
-s [<.tpr/.gro/...>] (topol.tpr) (Opt.)
Structure+mass(db): tpr gro g96 pdb brk ent
-n [<.ndx>] (index.ndx) (Opt.)
Index file
-fr [<.ndx>] (frames.ndx) (Opt.)
Index file
-sub [<.ndx>] (cluster.ndx) (Opt.)
Index file
-drop [<.xvg>] (drop.xvg) (Opt.)
xvgr/xmgr file
Options to specify output files:
-o [<.xtc/.trr/...>] (trajout.xtc)
Trajectory: xtc trr gro g96 pdb tng
Other options:
-b <time> (0)
Time of first frame to read from trajectory (default unit ps)
-e <time> (0)
Time of last frame to read from trajectory (default unit ps)
-tu <enum> (ps)
Unit for time values: fs, ps, ns, us, ms, s
-[no]w (no)
View output .xvg, .xpm, .eps and .pdb files
-xvg <enum> (xmgrace)
xvg plot formatting: xmgrace, xmgr, none
-skip <int> (1)
Only write every nr-th frame
-dt <time> (0)
Only write frame when t MOD dt = first time (ps)
-[no]round (no)
Round measurements to nearest picosecond
-dump <time> (-1)
Dump frame nearest specified time (ps)
-t0 <time> (0)
Starting time (ps) (default: don't change)
-timestep <time> (0)
Change time step between input frames (ps)
-pbc <enum> (none)
PBC treatment (see help text for full description): none, mol, res,
atom, nojump, cluster, whole
-ur <enum> (rect)
Unit-cell representation: rect, tric, compact
-[no]center (no)
Center atoms in box
-boxcenter <enum> (tric)
Center for -pbc and -center: tric, rect, zero
-box <vector> (0 0 0)
Size for new cubic box (default: read from input)
-trans <vector> (0 0 0)
All coordinates will be translated by trans. This can
advantageously be combined with -pbc mol -ur compact.
-shift <vector> (0 0 0)
All coordinates will be shifted by framenr*shift
-fit <enum> (none)
Fit molecule to ref structure in the structure file: none,
rot+trans, rotxy+transxy, translation, transxy, progressive
-ndec <int> (3)
Number of decimal places to write to .xtc output
-[no]vel (yes)
Read and write velocities if possible
-[no]force (no)
Read and write forces if possible
-trunc <time> (-1)
Truncate input trajectory file after this time (ps)
-exec <string>
Execute command for every output frame with the frame number as
argument
-split <time> (0)
Start writing new file when t MOD split = first time (ps)
-[no]sep (no)
Write each frame to a separate .gro, .g96 or .pdb file
-nzero <int> (0)
If the -sep flag is set, use these many digits for the file numbers
and prepend zeros as needed
-dropunder <real> (0)
Drop all frames below this value
-dropover <real> (0)
Drop all frames above this value
-[no]conect (no)
Add CONECT PDB records when writing .pdb files. Useful for
visualization of non-standard molecules, e.g. coarse grained ones.
Can only be done when a topology (tpr) file is present-s [<.tpr/.gro/...>] (topol.tpr) 输入模拟使用的.tpr文件
-f [<.xtc/.trr/...>] (traj.xtc) 输入模拟的轨迹文件
-o [<.xtc/.trr/...>] (trajout.xtc) 输出模拟后的轨迹文件
-pbc 设置周期性边界条件处理的类型
- mol 将分子的质心放在盒子中,并要求运行输入文件提供 -s
- res 将残基的质心放在框中。
- nojump 检查原子是否跳过盒子,然后将它们放回处。这具有所有分子将保持完整的效果(假设它们在初始构象中是完整的)。
- whole 会使破碎的分子成为整体。
-ur 设置 -pbc 的选项 mol、res 和 atom 的晶胞表示。
compact 将所有原子放在离盒子中心最近的地方。
接下来的RMSD分析
查看体系的稳定性。GROMACS内置的rms模块可用于计算RMSD.
gmxrms通过计算均方根偏差(RMSD)来比较两个结构root mean square deviation
均方根偏差,一般以第一帧的结构为参考结构,然后计算每一帧结构与第一帧结构的差异。选择4("Backbone")输出图将显示相对于最小化平衡系统中存在的结构的RMSD。计算相对于晶体结构的RMSD
gmx rms -s md_0_I.tpr -f md_0_I_noPBC.xtc -o rmsd.xvg -tu ns
gmx rms -s em.tpr -f md_0_I_noPBC.xtc -o rmsd_xtal.xvg -tu ns首先还是先看一下参数
Options to specify input files:
-s [<.tpr/.gro/...>] (topol.tpr)
Structure+mass(db): tpr gro g96 pdb brk ent
-f [<.xtc/.trr/...>] (traj.xtc)
Trajectory: xtc trr cpt gro g96 pdb tng
-f2 [<.xtc/.trr/...>] (traj.xtc) (Opt.)
Trajectory: xtc trr cpt gro g96 pdb tng
-n [<.ndx>] (index.ndx) (Opt.)
Index file
Options to specify output files:
-o [<.xvg>] (rmsd.xvg)
xvgr/xmgr file
-mir [<.xvg>] (rmsdmir.xvg) (Opt.)
xvgr/xmgr file
-a [<.xvg>] (avgrp.xvg) (Opt.)
xvgr/xmgr file
-dist [<.xvg>] (rmsd-dist.xvg) (Opt.)
xvgr/xmgr file
-m [<.xpm>] (rmsd.xpm) (Opt.)
X PixMap compatible matrix file
-bin [<.dat>] (rmsd.dat) (Opt.)
Generic data file
-bm [<.xpm>] (bond.xpm) (Opt.)
X PixMap compatible matrix file
Other options:
-b <time> (0)
Time of first frame to read from trajectory (default unit ps)
-e <time> (0)
Time of last frame to read from trajectory (default unit ps)
-dt <time> (0)
Only use frame when t MOD dt = first time (default unit ps)
-tu <enum> (ps)
Unit for time values: fs, ps, ns, us, ms, s
-[no]w (no)
View output .xvg, .xpm, .eps and .pdb files
-xvg <enum> (xmgrace)
xvg plot formatting: xmgrace, xmgr, none
-what <enum> (rmsd)
Structural difference measure: rmsd, rho, rhosc
-[no]pbc (yes)
PBC check
-fit <enum> (rot+trans)
Fit to reference structure: rot+trans, translation, none
-prev <int> (0)
Compare with previous frame
-[no]split (no)
Split graph where time is zero
-skip <int> (1)
Only write every nr-th frame to matrix
-skip2 <int> (1)
Only write every nr-th frame to matrix
-max <real> (-1)
Maximum level in comparison matrix
-min <real> (-1)
Minimum level in comparison matrix
-bmax <real> (-1)
Maximum level in bond angle matrix
-bmin <real> (-1)
Minimum level in bond angle matrix
-[no]mw (yes)
Use mass weighting for superposition
-nlevels <int> (80)
Number of levels in the matrices
-ng <int> (1)
Number of groups to compute RMS between-s [<.tpr/.gro/...>] (topol.tpr) 输入模拟使用的tpr文件
-f [<.xtc/.trr/...>] (traj.xtc) 输入处理周期性后的轨迹文件
-o [<.xvg>] (rmsd.xvg) 输出每一帧结构的rmsd,xvg文件
-tu <enum>(ps) 单位时间:fs, ps, ns, us, ms, s
-tu选项设定输出结果的时间单位为ns,即便轨迹文件以ps为单位输出。这是为了使输出文件更加清晰(尤其当模拟时间很长时,100 ns比起10^5 ps更美观)。
这个过程有两个选项,首先是拟合对齐 (Least Squares Fit) 怎么选?
这一步的目的是去除蛋白质的整体运动(平动和转动),只保留内部构象变化。
组名 | 原子数 | 是否推荐用于拟合? | 理由 |
|---|---|---|---|
C-alpha (3) | 129 | ✅ 非常推荐 | 每个残基一个,计算快,代表骨架走向。是计算RMSF和结构叠合的标准选择。 |
Backbone (4) | 387 | ✅ 推荐 | 包含CA、N、C,比只用C-α更严格,叠合更精确。是很多论文的默认选择。 |
Protein (1) | 1960 | ❌ 一般不用 | 包含柔性的侧链。用所有原子拟合,会使叠合“过度”匹配柔性区域,掩盖真正的骨架运动。 |
MainChain+Cb (6) | 634 | ⚠️ 有时可用 | 包含主链和Beta碳,比主链更稳定。如果体系中有大量柔性loop,这个选项也不错。 |
SideChain (8) | 1314 | ❌ 绝对不要用 | 侧链非常柔性,用它们做拟合会导致极其糟糕的叠合结果。 |
然后是RMSD 计算 怎么选?
在对齐之后,你可以选择同一组或不同组的原子来计算RMSD值。
组名 | 推荐用于计算? | 解释 |
|---|---|---|
C-alpha | ✅ 非常常见 | 给出蛋白质骨架的总体RMSD,对折叠/解折叠敏感。 |
Backbone | ✅ 非常常见 | 与C-α结果类似,但包含了肽键平面的信息,更全面。 |
Protein | ⚠️ 少用 | 包含侧链,RMSD值会比主链高,更能反映侧链的构象变化。如果想评估“全原子”稳定性可以用。 |
SideChain | 有时用 | 单独分析侧链的柔性。通常先做 Protein 减去 Backbone 来间接得到。 |
总结推荐
分析目的 | 推荐拟合组 (Least Squares Fit) | 推荐计算组 (RMSD Calculation) |
|---|---|---|
快速查看蛋白质稳定性 | C-alpha (3) | C-alpha (3) |
精密/发表级分析 | Backbone (4) | Backbone (4) 或 C-alpha (3) |
分析侧链柔性 | Backbone (4) 或 C-alpha (3) | Protein (1) |
分析活性中心局部变化 | Backbone (4) | 自定义的活性中心原子索引组 |
看一下分析出来的结果
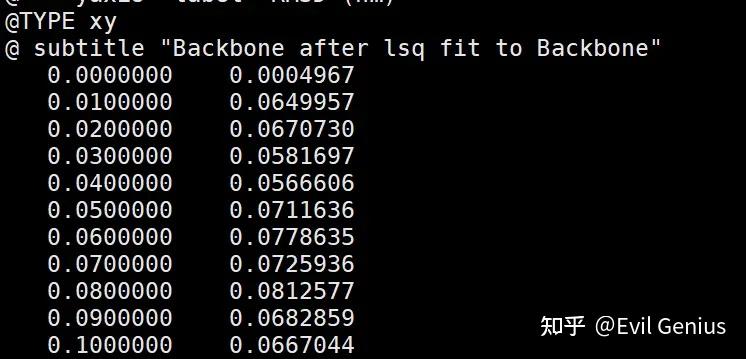
RMSD变化非常小,一般认为小于2Å的结果就非常好。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号