Nat. Biotechnol.|92个临床激酶抑制剂重做全景画像:副靶点,也可能是复用机会

Nat. Biotechnol.|92个临床激酶抑制剂重做全景画像:副靶点,也可能是复用机会

DrugAI
发布于 2026-04-28 10:28:07
发布于 2026-04-28 10:28:07
近日,Fred Hutchinson Cancer Center、Reaction Biology 和 University of Washington 等机构的研究团队在 Nature Biotechnology 发表了一项研究。论文由 Mehlam Saifudeen、Songli Zhu、Shuguang Liang 等共同第一作者完成,Marina Chan 和 Taranjit S. Gujral 为通讯作者。
这项工作做的事情很直接:把一批临床激酶抑制剂重新放到整个 kinome 里测了一遍。
激酶抑制剂是肿瘤靶向治疗中最重要的一类药物。很多药物通常会按照主靶点来理解,比如 BCR-ABL 抑制剂、EGFR 抑制剂、ALK 抑制剂、MET 抑制剂等。
但实际情况往往更复杂。很多激酶抑制剂并不只抑制一个靶点,它们还会作用于其他激酶。过去,这类作用常被视为 off-target 风险,因为它可能带来毒性和不可预测的副作用。
不过,从另一个角度看,副靶点也可能是药物复用机会。
这篇文章的核心,就是系统性地回答一个问题:
现有临床激酶抑制剂到底还能抑制哪些激酶?这些额外靶点里,有没有可以用于药物复用和精准治疗的线索?
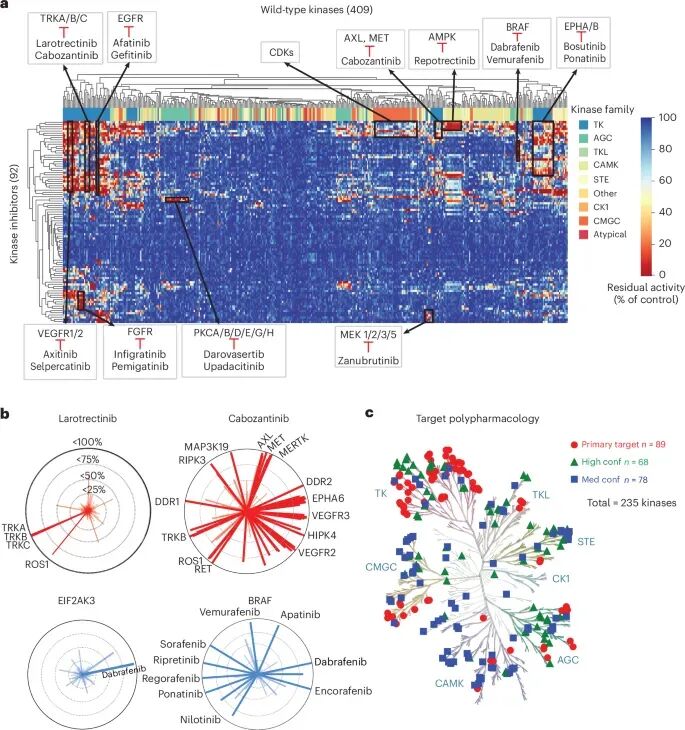
92个临床激酶抑制剂在409个野生型激酶上的抑制谱,以及从89个主靶点扩展到235个可抑制激酶的整体结果。
92个临床激酶抑制剂在409个野生型激酶上的抑制谱,以及从89个主靶点扩展到235个可抑制激酶的整体结果。
一句话概括
这篇文章通过系统性生化 profiling,把临床激酶抑制剂的作用范围从 89 个已知主靶点扩展到 235 个可被抑制的激酶,并进一步发现了多个突变特异性抑制和药物复用线索。
作者共测试了 92 个临床激酶抑制剂,其中 86 个为 FDA 已批准药物,另有 1 个由中国国家药监局批准,5 个处于后期临床阶段。
这些药物被系统测试在 758 个激酶 上,包括:
- 409 个野生型激酶
- 349 个肿瘤相关激酶变体
- 311 个突变体
- 38 个融合变体
最终,作者生成了约 29 万条 kinase–drug interaction measurements。
这不是一个单纯的筛选实验,更像是一张面向药物复用、突变耐药和精准肿瘤学的功能图谱。
重要性
临床上,很多激酶抑制剂是按照主靶点开发和使用的。药物标签会告诉研究者,它主要抑制哪个靶点,适用于哪类突变或适应症。
但药物真实的作用范围可能比标签更宽。
这在肿瘤治疗中尤其重要。肿瘤信号通路高度冗余,一个靶点被抑制后,细胞可能通过旁路通路继续生长。某些情况下,药物的多靶点性不一定是缺点,反而可能帮助同时压制一个信号网络。
问题是,过去缺少足够系统、定量、可比较的数据。
已有研究虽然做过激酶抑制剂 profiling,但很多主要集中在野生型激酶,对突变激酶和融合激酶的覆盖有限。而真实肿瘤治疗中,最关键的往往不是一个抽象的靶点,而是具体突变背景下的靶点。
因此,这篇文章的价值在于:它把临床激酶抑制剂、野生型激酶、肿瘤突变变体和药物复用线索放到同一个实验框架中比较。
这项研究怎么做
作者使用的是 Reaction Biology 的 HotSpot radiometric filter-binding kinase assay。
这个 assay 直接测量激酶把放射性标记磷酸从 ATP 转移到底物蛋白或肽段上的过程。因此,它检测的是激酶催化活性的变化,而不只是药物和蛋白之间的结合。
所有药物在完整 panel 中以 1 μM 浓度进行重复检测,并在 Km ATP 条件下完成。作者在正式筛选前做了剂量反应实验和统计分析,用来支持 1 μM 作为统一筛选浓度。
需要注意的是,1 μM 主要是为了建立统一的生化参考图谱,并不等同于临床暴露水平。也就是说,一个药物在这个条件下抑制某个激酶,并不代表它一定能在患者体内达到同样效果。
这也是整篇文章需要谨慎理解的地方:这是一张高价值的假设生成图谱,不是直接的临床用药结论。
从89个主靶点,到235个可抑制激酶
在这批药物中,原本标注或设计的 primary kinase targets 一共有 89 个。
但通过系统 profiling,作者发现现有临床激酶抑制剂还能抑制另外 146 个激酶。换句话说,这些药物覆盖的激酶空间从 89 个主靶点扩展到 235 个激酶。
这也是文章最核心的发现之一。
过去很多作用可能被简单归为 off-target,现在有了更系统的数据,可以进一步判断它们到底是毒性风险,还是复用机会。
作者还用 KISS score、Gini coefficient 和 CATDS score 等指标评估药物选择性。比如,TRK 抑制剂 larotrectinib 表现出很高选择性,而 gilteritinib 的抑制谱更广,提示其作用范围比 FLT3 标签靶点更宽。
案例一:tepotinib 可能复用为 IRAK1/4 抑制剂
第一个验证案例是 tepotinib 和 IRAK1/4。
IRAK1 和 IRAK4 参与 Toll-like receptor 和 interleukin-1 signaling,在先天免疫和炎症调控中发挥作用。文中指出,IRAK1/4 与多种癌症、代谢和炎症疾病相关,但目前没有 FDA 批准的 IRAK1/4 抑制剂。
在 profiling 数据中,作者发现 tepotinib 可以抑制 IRAK1 和 IRAK4。随后,研究团队用 NanoBRET 和 CETSA 验证了 tepotinib 与 IRAK4,以及细胞内 IRAK1/4 的结合或稳定化证据。
接下来,作者把验证重点放在 glioma 模型中。TCGA 数据显示,高 IRAK1/4 表达与 glioblastoma 患者较差的总体生存相关。细胞实验中,tepotinib 能降低多个 glioma 模型的 neurosphere growth。
转录组分析进一步显示,tepotinib 处理后,胆固醇外排相关基因 ABCA1 和 ABCG1 上调,同时 NF-κB、SREBF2、steroid biosynthesis 和 cholesterol synthesis 相关通路下降。小鼠 PDX 模型中,tepotinib 单药处理也抑制了肿瘤生长。
这个案例说明,一个原本作为 MET 抑制剂使用的药物,可能通过 IRAK1/4 相关机制在 glioma 中产生新的复用机会。
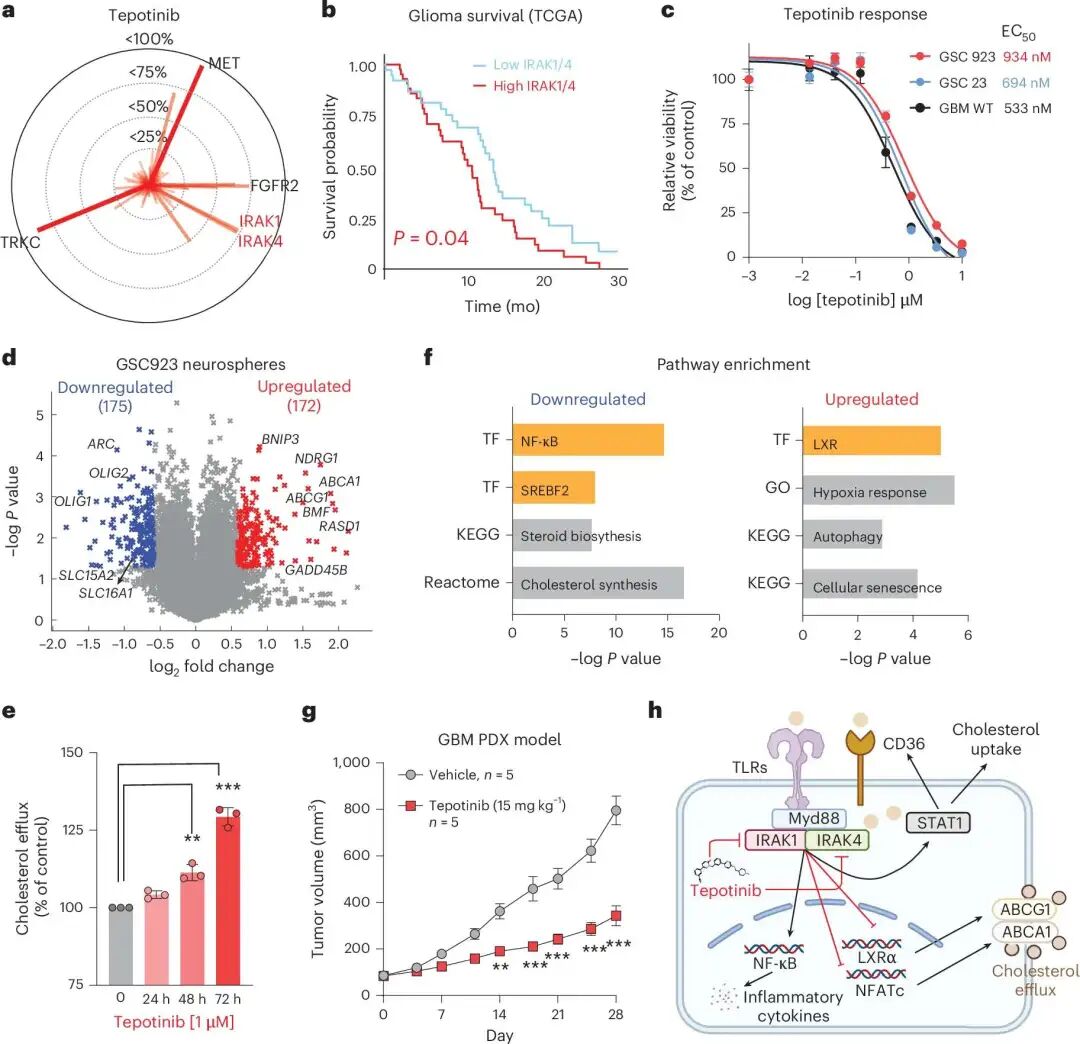
tepotinib 抑制 IRAK1/4,并在 glioma 模型中影响胆固醇代谢、炎症信号和肿瘤生长。
tepotinib 抑制 IRAK1/4,并在 glioma 模型中影响胆固醇代谢、炎症信号和肿瘤生长。
案例二:gilteritinib 影响 EMT,并增强 MEK 抑制剂敏感性
第二个案例与 EMT 相关。
EMT,也就是 epithelial–mesenchymal transition,是肿瘤侵袭、转移和治疗耐受中的重要过程。作者利用 CCLE 数据比较了 mesenchymal-like 和 epithelial-like 癌细胞系中的激酶表达,识别出一批与 mesenchymal 状态相关的激酶。
在这批 mesenchymal-associated kinases 上,gilteritinib、pacritinib 和 midostaurin 表现出较广的抑制谱。其中,gilteritinib 能抑制 26 个 mesenchymal-associated kinases。
在 MDA-MB-231 细胞中,gilteritinib 降低了 EMT marker vimentin 和 slug,同时增加 epithelial marker E-cadherin。转录组结果也显示,gilteritinib 处理后,下调基因富集于 ECM organization、collagen assembly 和 PI3K–AKT pathway 等与迁移和生长相关的过程。
功能实验中,gilteritinib 还能剂量依赖性降低细胞迁移。更重要的是,它增强了细胞对 MEK 抑制剂 cobimetinib 的敏感性,使 cobimetinib 的 EC50 从 1600 nM 降至 46 nM。
这里的重点不是简单说 gilteritinib 可以治疗某种癌症,而是说明:
广谱激酶抑制剂的多靶点作用,可能被用来干预 EMT 这类复杂细胞状态。
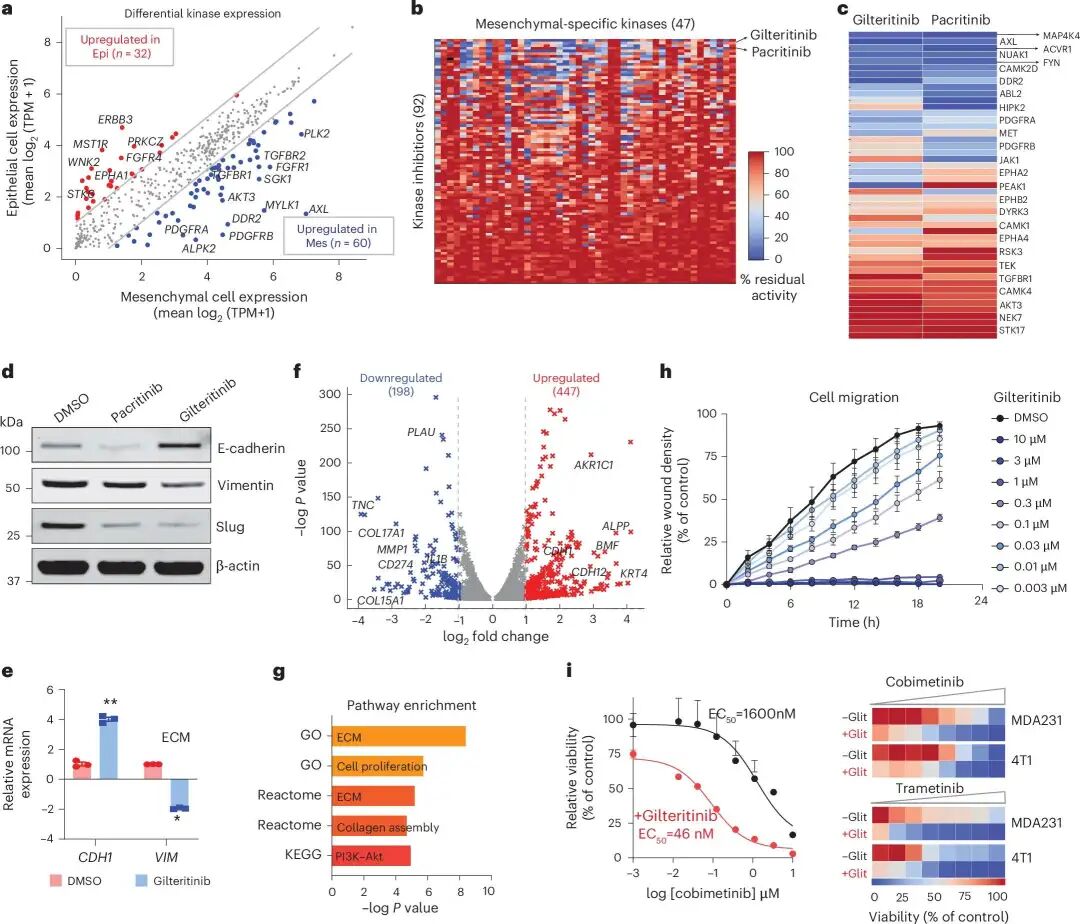
gilteritinib 靶向 mesenchymal-associated kinases,降低 EMT 特征,并增强乳腺癌细胞对 MEK 抑制剂的敏感性。
gilteritinib 靶向 mesenchymal-associated kinases,降低 EMT 特征,并增强乳腺癌细胞对 MEK 抑制剂的敏感性。
案例三:brigatinib 抑制 MARK2/3,影响 Hippo–YAP signaling
第三个案例关注 kinase paralogs。
很多激酶存在 paralog,功能有重叠。单独敲除一个基因时,另一个 paralog 可能补偿其功能,因此 CRISPR 单基因依赖性筛选未必能完整反映这类靶点的可药性。
作者系统分析了临床激酶抑制剂对 paralogous kinase families 的覆盖,并重点验证了 MARK family。
MARK kinases 与细胞极性、分化、微管动态和细胞分裂有关。已有研究提示,MARK2 和 MARK3 在 YAP-driven tumors 中可能是 codependencies。
在 KiRHub 数据中,midostaurin 和 brigatinib 对 MARK family kinases 有抑制作用。作者选择 brigatinib 做进一步验证。
在 Panc1 胰腺癌细胞中,brigatinib 处理后,Hippo–YAP signaling 相关基因如 CYR61、CTGF、AMOTL2、SERPINE1、TGFB2 和 WNT5B 下调。通路分析也显示,Hippo signaling、MAPK signaling 和 collagen assembly 等通路受到影响。
在 Panc1 xenograft 模型中,brigatinib 单药处理降低了肿瘤生长和肿瘤重量。
这个案例提示,系统性 paralog mapping 可以帮助发现已有药物对冗余激酶网络的覆盖能力,尤其适合分析 YAP-driven cancer 这类涉及通路补偿的场景。
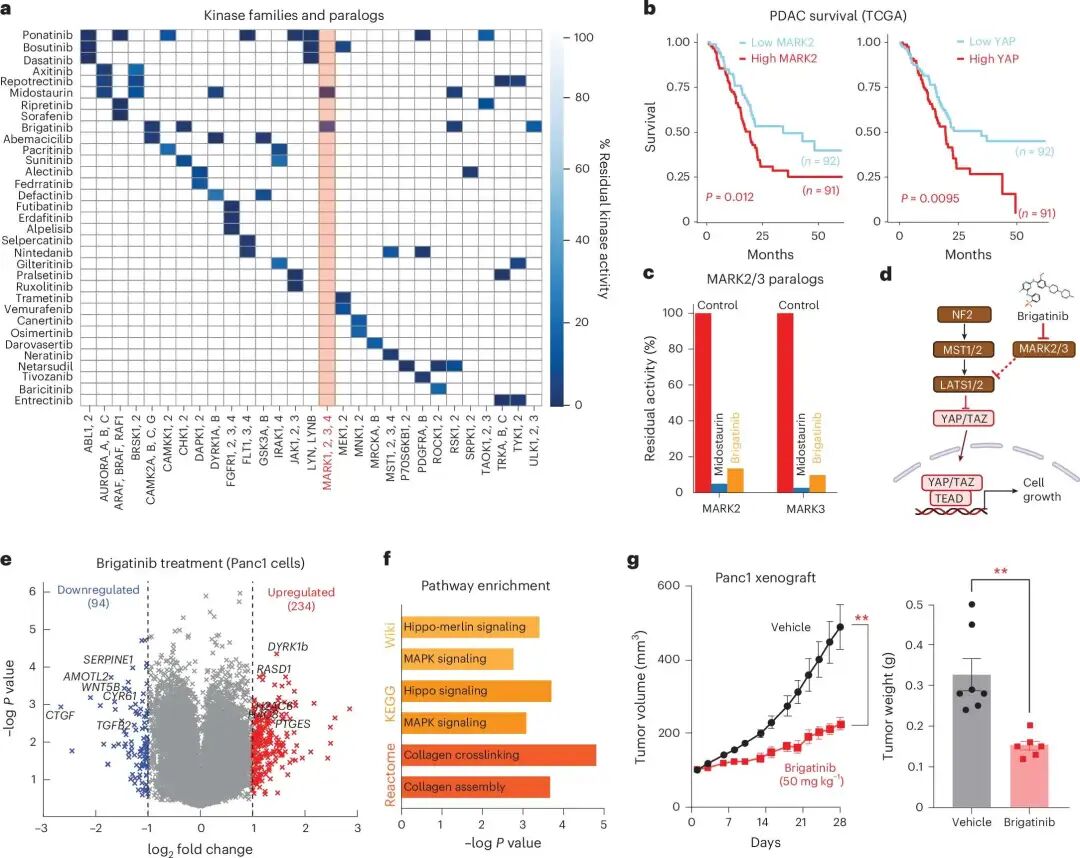
brigatinib 抑制 MARK2/3,并在 pancreatic cancer 模型中影响 Hippo–YAP signaling 和肿瘤生长。
brigatinib 抑制 MARK2/3,并在 pancreatic cancer 模型中影响 Hippo–YAP signaling 和肿瘤生长。
突变和融合变体:同一个靶点,不同突变可能完全不同
这篇文章另一个重要部分,是对 349 个激酶变体 的系统 profiling。
其中包括:
- 311 个 kinase mutations
- 38 个 kinase gene fusions
结果显示,在该实验阈值下,293/311 个突变体,即 94% ,能被至少一个现有临床激酶抑制剂抑制超过 70%;37/38 个融合变体,即 97% ,也能被至少一个药物抑制超过 70%。
这个结果说明,现有激酶抑制剂对临床相关 kinase variants 的覆盖范围,可能比药物标签暗示的更广。
但更关键的是,同一个激酶的不同突变,对药物的敏感性可能差异很大。
作者以 ALK、EGFR、RET、FGFR 和 MET 等为例,展示了突变特异性的药物敏感性。对于精准治疗来说,这一点非常重要。因为真实临床用药面对的不是抽象的 EGFR 或 MET,而是某个具体突变背景下的 EGFR 或 MET。
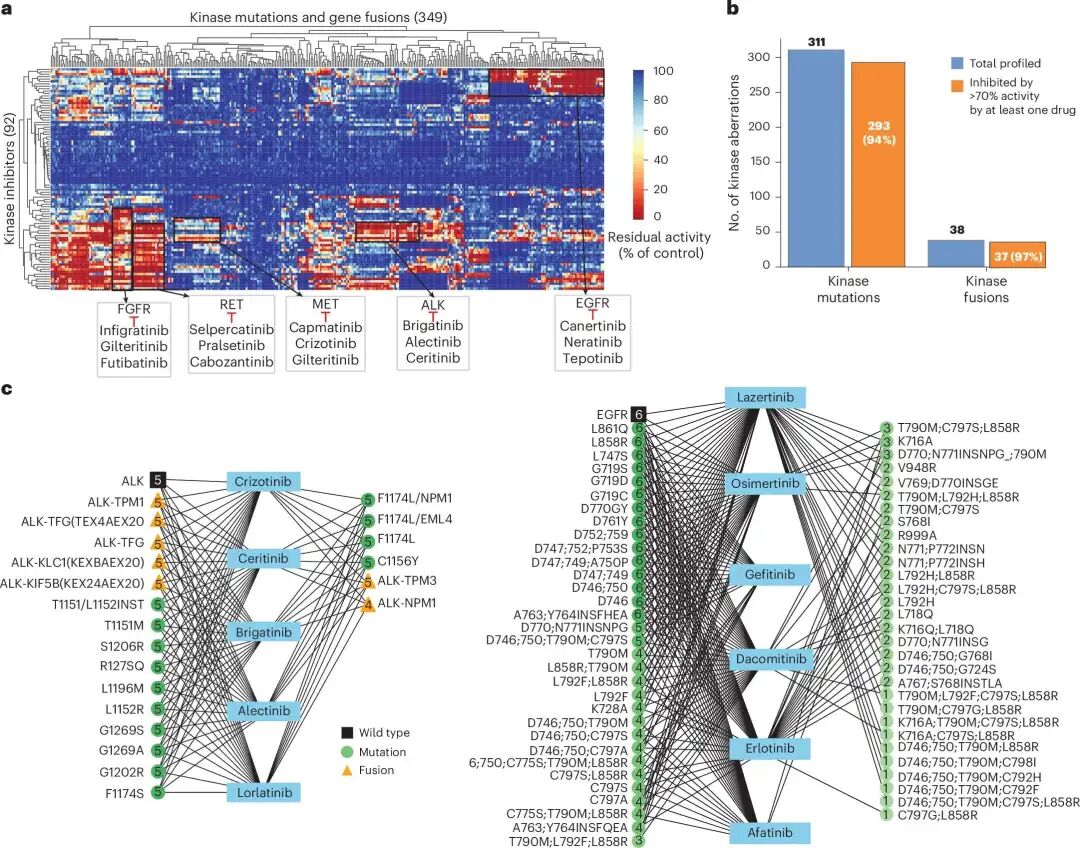
92个临床激酶抑制剂对349个突变和融合激酶变体的抑制谱,显示出明显的 variant-specific drug selectivity。
92个临床激酶抑制剂对349个突变和融合激酶变体的抑制谱,显示出明显的 variant-specific drug selectivity。
FGFR 和 MET:从耐药突变里寻找复用机会
为了展示这个数据集如何发现治疗缺口,作者进一步分析了 FGFR 和 MET。
在 FGFR family 中,作者发现有 6 个 FGFR mutants 对 futibatinib、pemigatinib 和 infigratinib 这三种 FGFR 抑制剂均不敏感。随后,研究团队扩大搜索范围,发现 pralsetinib、selpercatinib 和 erdafitinib 对部分 resistant FGFR variants 具有活性。
在 MET 部分,作者发现 4 个位于 D1228 残基的 MET mutants 对已批准 MET 抑制剂无效,另有 3 个 mutants 仅被 cabozantinib 抑制。但 profiling 数据显示,这 7 个 MET variants 均对 gilteritinib 敏感。
进一步实验显示,gilteritinib 对 MET-D1228A 表达细胞具有较强抑制活性,文中报告的 EC50 为 0.21 μM,优于所比较的已批准 MET 抑制剂范围 1.2–10 μM。CETSA 实验也支持 gilteritinib 与 MET-Y1230H 的细胞内 target engagement。
这里的重点不是说 gilteritinib 立刻可以用于 MET 突变患者,而是说明:
mutation-specific profiling 可以帮助识别现有药物覆盖不到的突变,并从已有药物中寻找候选复用方案。
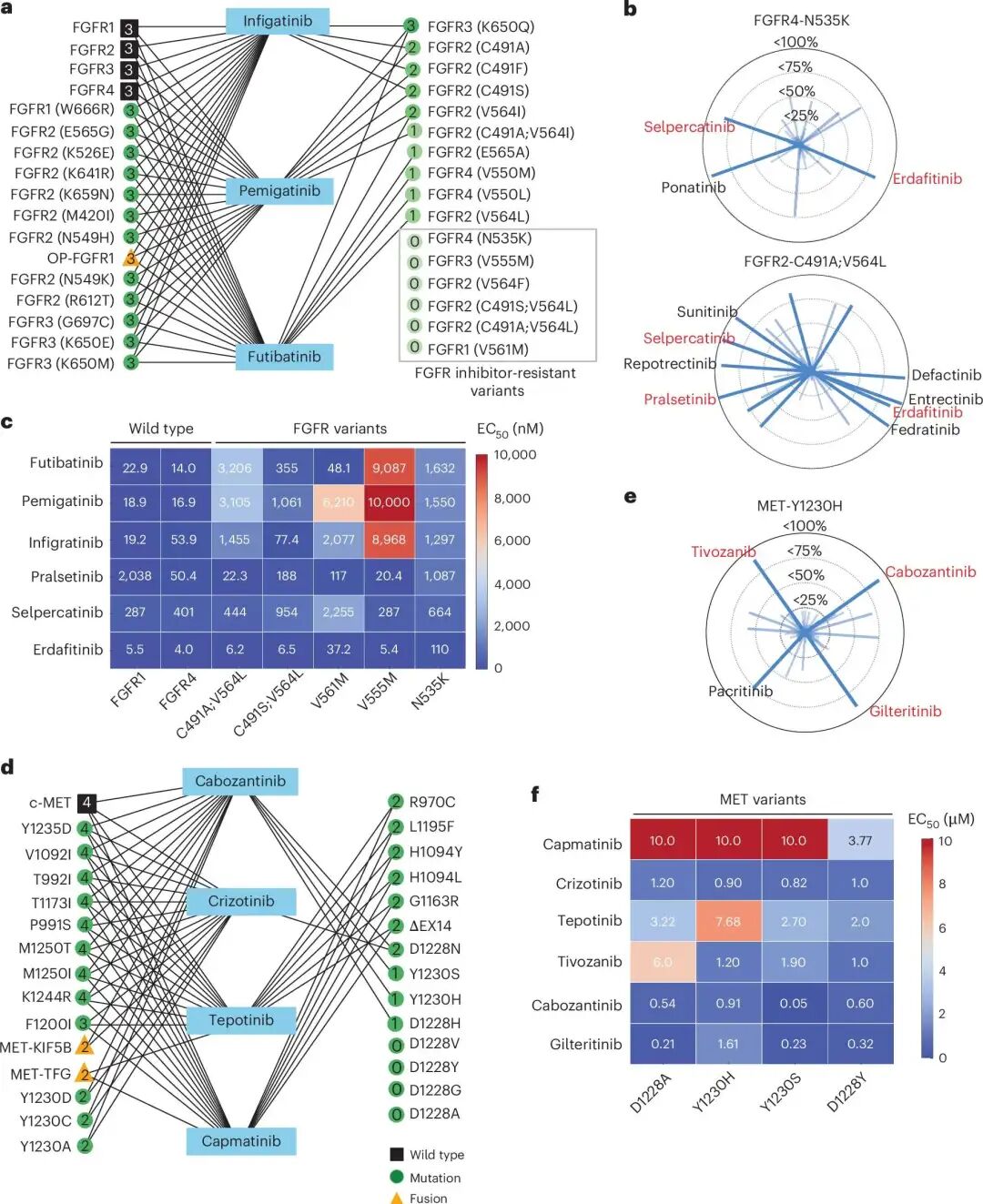
FGFR 和 MET 突变中的治疗缺口与复用机会。图中展示了不同 FGFR 和 MET variants 对现有临床激酶抑制剂的差异性敏感性。
FGFR 和 MET 突变中的治疗缺口与复用机会。图中展示了不同 FGFR 和 MET variants 对现有临床激酶抑制剂的差异性敏感性。
KIRHub:把数据变成可查询资源
为了方便研究者使用这批数据,作者开发了一个网页工具:KIRHub,Kinase Inhibitor Repurposing Hub。
https://kirhub.fredhutch.org/
KIRHub 主要支持三类查询:
- 寻找可以抑制 mutant kinases 的药物
- 寻找可以抑制 wild-type kinases 的药物
- 探索不同癌症 lineage 中的 kinase essentiality
这个工具的价值在于,它把一张复杂的 kinase–drug interaction matrix 变成了可以搜索、排序和下载的资源。
对做药物复用、激酶信号通路、耐药突变和精准肿瘤学的人来说,这类资源可以直接用来生成假设。
对 AIDD 和药物复用的启发
这篇文章并不是一篇典型的 AI 药物设计论文,但对 AIDD 很有启发。
很多 AIDD 工作习惯从模型出发:预测活性、生成分子、做虚拟筛选,或者寻找新的化学结构。但药物发现中还有另一类问题同样重要:
已有药物到底还能做什么?
这篇文章给出的答案是,药物复用不能只靠药物标签、文献印象或简单相似性判断,而需要系统、定量、可比较的靶点谱数据。
一个有价值的复用线索,至少需要同时回答几个问题:
- 这个药物是否真的抑制目标 kinase?
- 这种抑制是否发生在具体突变背景下?
- 相关癌症是否依赖这个 kinase 或信号网络?
- 细胞和动物模型中是否能看到功能效应?
- 药物浓度、暴露和安全性是否可能支持进一步验证?
这也是这篇文章对 AIDD 的提醒:真正有价值的药物复用,不只是找到一个 drug–target pair,而是把这个连接放回具体突变、具体细胞状态和具体疾病依赖性中验证。
需要谨慎看待的地方
这项工作数据规模很大,但边界也很清楚。
首先,主要 profiling 是在纯化 kinase constructs 和标准化 assay 条件下完成的。这有利于跨药物、跨激酶比较,但不能完整模拟细胞内环境。
其次,HotSpot assay 主要测量 catalytic kinase inhibition。对于依赖调控结构域、蛋白复合物、亚细胞定位或非催化功能的机制,这类 assay 未必能完整反映。
第三,PI3K 这类 lipid kinases 没有被纳入 profiling,因为该 assay 平台不适合测量脂质激酶活性。
第四,1 μM 筛选浓度是为了建立统一的 biochemical reference,并不等同于临床暴露水平。一个药物在 1 μM 下抑制某个 kinase,并不自动说明它在患者体内能够产生足够 target engagement。
因此,KIRHub 更适合被理解为 高价值的假设生成资源,而不是临床用药指南。
小结
这篇 Nature Biotechnology 文章重新绘制了临床激酶抑制剂的作用版图。
作者用统一的生化活性检测体系,系统评估了 92 个临床激酶抑制剂对 758 个激酶的抑制作用,生成约 29 万条 kinase–drug interaction measurements。结果显示,现有临床激酶抑制剂覆盖的激酶空间远大于主靶点标注:可被抑制的激酶从 89 个 primary targets 扩展到 235 个。
更重要的是,作者不仅分析了野生型激酶,还覆盖了 349 个突变和融合变体,揭示了广泛的 mutation-specific selectivity。FGFR 和 MET 案例显示,同一个靶点的不同突变可能对药物表现出完全不同的敏感性,而已有药物中也可能存在绕开耐药突变的复用机会。
这项工作的价值不在于证明某个药物马上可以用于某个新适应症,而在于提供了一张可查询、可验证、可继续扩展的功能性激酶抑制剂地图。
对于药物复用和精准肿瘤学而言,这类系统性 profiling 资源可能比单个新模型更接近真实决策需求:它不直接给出临床答案,但能告诉我们,哪些已有药物、哪些突变、哪些通路,值得被进一步认真验证。
论文: Comprehensive profiling of clinically approved kinase inhibitors reveals mutation-specific inhibitors and opportunities for drug repurposing
期刊: Nature Biotechnology,2026
DOI: 10.1038/s41587-026-03090-8
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-24,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号