分析梳理--分子动力学模拟的常规步骤三(Gromacs)
原创
分析梳理--分子动力学模拟的常规步骤三(Gromacs)
原创
追风少年i
发布于 2026-04-21 14:53:00
发布于 2026-04-21 14:53:00
作者,Evil Genius
今天我们继续分子动力学:平衡电荷。
前面的过程我们设置了溶剂盒子并添加溶剂,生成了solv.gro文件。
这个过程分两步走。
第一步:gmx grompp。
gmx grompp (the gromacs preprocessor)读取分子拓扑文件,检查文件的有效性,将拓扑从分子描述扩展为原子描述。拓扑文件包含有关分子类型和分子数量的信息,预处理器根据需要复制每个分子。分子类型的数量没有限制。键和键角可以转换为约束,分别用于氢和重原子。然后读取坐标文件,如果需要,可以从麦克斯韦分布生成速度。gmx grompp还读取gmx mdrun的参数(例如MD步数、时间步长、截止值)和其他参数,例如NEMD参数,这些参数经过校正以使净加速度为零。一个二进制文件,可以作为MD程序的唯一输入文件。
我们还是来看看参数
-f [<.mdp>] (grompp.mdp)
grompp input file with MD parameters
-c [<.gro/.g96/...>] (conf.gro)
Structure file: gro g96 pdb brk ent esp tpr
-r [<.gro/.g96/...>] (restraint.gro) (Opt.)
Structure file: gro g96 pdb brk ent esp tpr
-rb [<.gro/.g96/...>] (restraint.gro) (Opt.)
Structure file: gro g96 pdb brk ent esp tpr
-n [<.ndx>] (index.ndx) (Opt.)
Index file
-p [<.top>] (topol.top)
Topology file
-t [<.trr/.cpt/...>] (traj.trr) (Opt.)
Full precision trajectory: trr cpt tng
-e [<.edr>] (ener.edr) (Opt.)
Energy file
-qmi [<.inp>] (topol-qmmm.inp) (Opt.)
Input file for QM program
Options to specify input/output files:
-ref [<.trr/.cpt/...>] (rotref.trr) (Opt.)
Full precision trajectory: trr cpt tng
Options to specify output files:
-po [<.mdp>] (mdout.mdp)
grompp input file with MD parameters
-pp [<.top>] (processed.top) (Opt.)
Topology file
-o [<.tpr>] (topol.tpr)
Portable xdr run input file
-imd [<.gro>] (imdgroup.gro) (Opt.)
Coordinate file in Gromos-87 format
Other options:
-[no]v (no)
Be loud and noisy
-time <real> (-1)
Take frame at or first after this time.
-[no]rmvsbds (yes)
Remove constant bonded interactions with virtual sites
-maxwarn <int> (0)
Number of allowed warnings during input processing. Not for normal
use and may generate unstable systems
-[no]zero (no)
Set parameters for bonded interactions without defaults to zero
instead of generating an error
-[no]renum (yes)
Renumber atomtypes and minimize number of atomtypes-f [<.mdp>] (grompp.mdp)输入文件,grompp的MD参数输入文件
-c[<.grol.g96/...>] (conf.gro)指定输入的结构文件-P.top>拓扑文件
-o[<.tpr>] (topol.tpr)该文件包含模拟的起始结构、分子拓扑结构和所有模拟参数。由于此文件是二进制格式,因此无法使用普通编辑器读取。
-maxwarn允许的警告数
其中的em.mdp文件
为了用grompp产生.tpr文件,我们还需要一个扩展名为.mdp(molecular dynamics parameter)的输入文件。grompp会将坐标和拓扑信息与.mdp文件中设定的参数组合起来生成.tpr文件。
.mdp文件通常用于运行能量最小化(EM)或分子动力学模拟(MD),但在这里我们只是简单地用它来生成系统的原子描述。一个.mdp文件的示例(后面我们将使用它)如下。

这里给大家一个em.mdp的文件范例
; md.mdp - 分子动力学模拟参数文件
; 预处理器
cpp = /usr/bin/cpp
define = -DPOSRES ; 如需位置限制,取消注释
; 运行控制
integrator = md ; 分子动力学积分器
tinit = 0 ; 起始时间 (ps)
dt = 0.002 ; 时间步长 (ps) = 2 fs
nsteps = 5000000 ; 总步数 (5M * 2 fs = 10 ns)
comm-mode = Linear
nstcomm = 100 ; 质心运动移除频率
; 邻居搜索与短程相互作用
cutoff-scheme = Verlet ; 适用于GPU加速
nstlist = 20 ; 邻居列表更新频率
ns-type = grid ; 网格搜索算法
pbc = xyz ; 周期性边界条件
rlist = 1.0 ; 邻居列表截断距离 (nm)
; 静电相互作用
coulombtype = PME ; 粒子网格Ewald方法
rcoulomb = 1.0 ; 静电截断距离 (nm)
pme-order = 4 ; PME插值阶数
fourierspacing = 0.16 ; PME网格间距 (nm)
; 范德华相互作用
vdwtype = Cut-off ; 截断类型
rvdw = 1.0 ; 范德华截断距离 (nm)
DispCorr = EnerPres ; 对能量/压力进行色散校正
; 温度耦合
tcoupl = V-rescale ; 速度重标定恒温器
tc-grps = Protein Non-Protein; 温度耦合组
tau_t = 0.1 0.1 ; 时间常数 (ps)
ref_t = 298 298 ; 参考温度 (K)
; 压力耦合
pcoupl = C-rescale ; 恒压器(也可用 Parrinello-Rahman)
pcoupltype = isotropic ; 各向同性压力耦合
tau_p = 2.0 ; 压力时间常数 (ps)
ref_p = 1.0 ; 参考压力 (bar)
compressibility = 4.5e-5 ; 压缩系数 (bar^-1)
; 约束算法
constraints = h-bonds ; 约束氢键(可用 all-bonds)
constraint-algorithm= LINCS ; 约束算法
lincs-order = 4 ; LINCS阶数
lincs-iter = 1 ; LINCS迭代次数
; 输出频率
nstxout = 0 ; 不输出坐标文件 (旧格式)
nstvout = 0 ; 不输出速度
nstfout = 0 ; 不输出力
nstxout-compressed = 1000 ; 每1000步输出压缩轨迹 (xtc)
nstenergy = 1000 ; 每1000步输出能量文件 (edr)
nstlog = 1000 ; 每1000步输出日志
; 其他选项
continuation = no ; 从头开始模拟(非继续)
gen-vel = yes ; 生成初速度
gen-temp = 298 ; 初速度对应温度
gen-seed = -1 ; 随机种子(-1 = 基于时间)然后我们运行
gmx grompp -f em.mdp -c solv.gro -p topol.top -o ions.tpr -maxwarn 3 -r solv.gro生成ions.tpr文件
接下来第二步:gmx genion
gmx genion用单原子离子随机替换溶剂分子。溶剂分子组应该是连续的,并且所有分子应该具有相同的原子数。应将离子分子添加到拓扑文件中或使用-p选项自动修改拓扑。
看一看全部参数
Options to specify input files:
-s [<.tpr>] (topol.tpr)
Portable xdr run input file
-n [<.ndx>] (index.ndx) (Opt.)
Index file
Options to specify input/output files:
-p [<.top>] (topol.top) (Opt.)
Topology file
Options to specify output files:
-o [<.gro/.g96/...>] (out.gro)
Structure file: gro g96 pdb brk ent esp
Other options:
-np <int> (0)
Number of positive ions
-pname <string> (NA)
Name of the positive ion
-pq <int> (1)
Charge of the positive ion
-nn <int> (0)
Number of negative ions
-nname <string> (CL)
Name of the negative ion
-nq <int> (-1)
Charge of the negative ion
-rmin <real> (0.6)
Minimum distance between ions and non-solvent
-seed <int> (0)
Seed for random number generator (0 means generate)
-conc <real> (0)
Specify salt concentration (mol/liter). This will add sufficient
ions to reach up to the specified concentration as computed from
the volume of the cell in the input .tpr file. Overrides the -np
and -nn options.
-[no]neutral (no)
This option will add enough ions to neutralize the system. These
ions are added on top of those specified with -np/-nn or -conc.-s[<.tpr>] (topol.tpr)上一步生成的tpr文件
-P[<.top>] (topol.top) (Optional)指定需要修改的拓扑文件
-o [<.grol.g96/...>] (out.gro)输出的结构文件
-pname <string>(NA)阳离子的名称
-nname <string>(CL)阴离子的名称
-np<int>(0)添加的阳离子的数量
-nn<int>(0)添加的阴离子的数量
拓展:
-conc<real>(0)指定盐浓度(摩尔/升)。 这将添加足够的离子以达到根据输入.tpr文件中的池体积计算得出的指定浓度。覆盖-np和-nn选项
-[no]neutral (no)此选项将添加足够的离子以中和系统。这些离子被添加到使用-np/-nn或-conc指定的离子之上。
慎用!!!
我们来运行一下
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -nn 8选择连续的溶剂分子组来替换成离子
Select a continuous group of solvent molecules
Group 0 ( System) has 33892 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31932 elements
Group 12 ( Water) has 31932 elements
Group 13 ( SOL) has 31932 elements
Group 14 ( non-Water) has 1960 elements
Select a group: 13
Selected 13: 'SOL'
Number of (3-atomic) solvent molecules: 10644最后在文件中添加了CL离子。
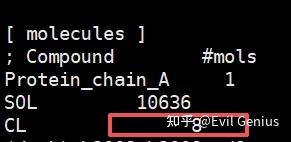
看一下生成的solv_ions.gro文件
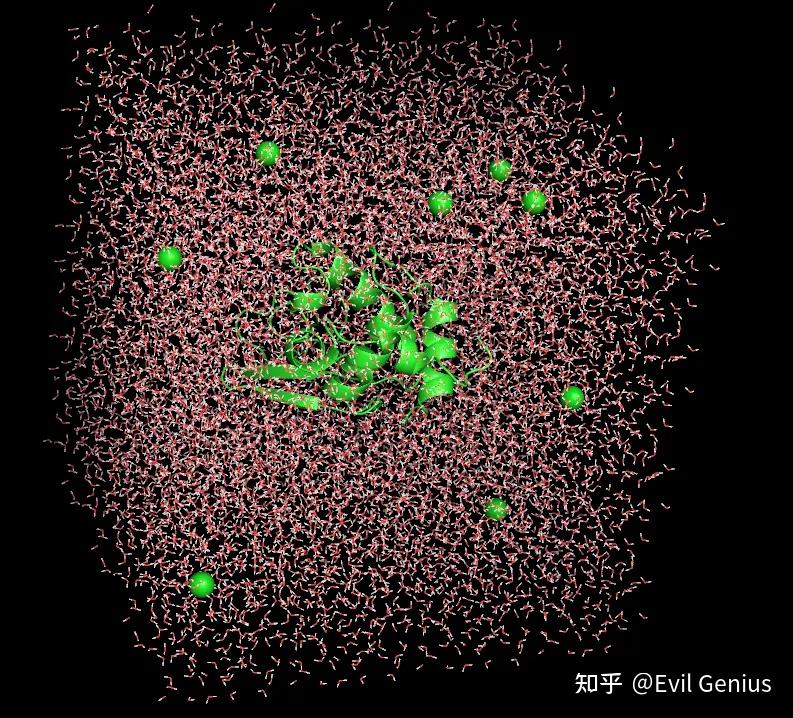
添加了CL离子,平衡电荷。
现在,已经添加了溶剂分子和离子,得到了一个电中性的体系.在开始动力学模拟之前,必须保证体系的结构正常,原子之间的距离不会过近,几何构型合理.对结构进行弛豫可以达到这些要求,这个过程称为能量最小化(EM,energyminimization).
下一篇我们分享。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号