ATMOS:基于状态空间模型的生物分子动力学原子轨迹 AI生成框架

ATMOS:基于状态空间模型的生物分子动力学原子轨迹 AI生成框架

DrugIntel
发布于 2026-04-21 11:08:34
发布于 2026-04-21 11:08:34

论文信息:Atomic Trajectory Modeling with State Space Models for Biomolecular Dynamics 作者:Liang Shi, Jiarui Lu, Junqi Liu, Chence Shi, Zhi Yang, Jian Tang 等 机构:北京大学计算机学院 · BioGeometry · Mila(魁北克 AI 研究所)· HEC Montréal 预印版:arXiv:2603.17633v1,2026 年 3 月 18 日
一、研究背景与动机
1.1 分子动力学模拟的核心价值与瓶颈
生物大分子(蛋白质、核酸、小分子配体)在生理条件下并非静态结构,而是处于持续的热力学运动之中。这种动力学行为决定了蛋白质的构象变化、酶的催化机制、受体的信号转导以及药物-靶标的结合过程。传统分子动力学(Molecular Dynamics, MD)模拟通过数值积分牛顿运动方程,在原子精度上重现这些物理过程,是研究生物分子动力学的"金标准"。
然而,MD 模拟面临严峻的计算效率瓶颈:
- • 时间尺度鸿沟:生物学相关的构象转变(如蛋白质折叠、受体激活)通常发生在微秒至毫秒量级,而 MD 的积分步长仅为飞秒( s),两者相差约 9 个数量级;
- • 计算资源消耗:对一个含有数万原子的系统模拟 1 μs 轨迹,即便使用专用硬件(如 Anton 超算)也需要数天;
- • 稀有事件采样困难:高能量垒分隔的构象转变在标准 MD 中极少自发发生,导致采样效率极低。
1.2 深度生成模型的现有路径及局限
为缓解上述计算负担,近年来深度生成模型被引入分子动力学加速领域,大致形成两条技术路线:
路线一:构象集成生成(Ensemble Generation)
代表工作包括 AlphaFlow(Jing et al., 2024)、BioEmu(Lewis et al., 2025)等。这类方法将问题建模为从玻尔兹曼平衡分布中采样,生成一组热力学上合理的独立构象。核心局限在于:生成的样本相互独立(i.i.d.),完全丢弃了时间维度,无法描述动力学路径、转变速率及构象演化轨迹。
路线二:轨迹生成(Trajectory Generation)
代表工作包括 MDGen(Jing et al., 2024)、TEMPO(Xu et al., 2025)、ConfRover(Shen et al., 2025)等。这类方法尝试建模时序演化,但存在两个关键限制:
- 1. 系统范围受限:大多数方法仅支持单体蛋白质(monomer protein),无法处理蛋白质-配体等复杂多分子体系;
- 2. 计算复杂度高:基于 Transformer 的自回归方法需要在每步预测时回顾全部历史,导致推理复杂度随轨迹长度二次增长()。
1.3 ATMOS 的定位
ATMOS(ATomic Trajectory MOdeling with SSMs)旨在同时解决上述两类方法的不足:以状态空间模型(SSM)为核心,在保留时序信息的同时,将系统范围从单体蛋白质扩展至蛋白质-配体复合物,并将推理复杂度降低至线性。
二、问题形式化
2.1 任务定义
设生物分子系统包含 个原子,其动力学轨迹表示为:
其中 为 时刻所有原子的三维笛卡尔坐标, 为轨迹帧数。时间不变的原子特征 包含原子类型、残基类型、分子身份(蛋白质/配体)等信息。
学习目标是拟合条件生成分布 ,按时间维度自回归分解:
2.2 状态空间模型(SSM)基础
SSM 提供了一个通用的序列建模框架。连续时间线性 SSM 通过如下常微分方程定义:
其中 ,, 为可学习投影矩阵。对离散序列(如以固定时间间隔 采样的 MD 轨迹),采用零阶保持(ZOH)方法离散化:
递归形式下每步推理复杂度为 ,相比 Transformer 的 具有显著优势,适合生成任意长度的分子轨迹。
物理直觉:分子动力学本质上满足马尔可夫性质——系统未来状态由当前相空间状态(位置 + 动量)决定,而非完整历史。SSM 的递归隐状态 自然对应这一相空间描述,隐式编码了动量、作用力以及恒温器状态等动力学变量,从物理层面与 MD 积分器形成对应。
三、模型架构
ATMOS 整体框架由三个功能模块串联构成(见图 1):几何编码器 → SSM 状态转移模块 → 扩散解码器。
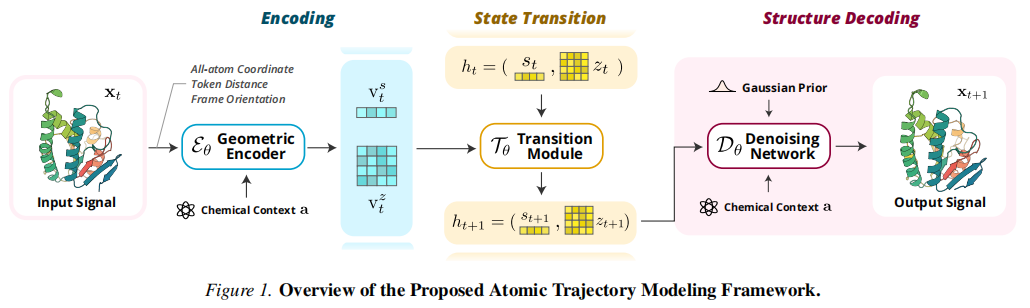
3.1 紧凑耦合隐变量表示
直接在全原子坐标空间建模大体系的交互在计算上不可行。ATMOS 借鉴 AlphaFold3(Abramson et al., 2024)的设计,将原子坐标"令牌化"为长度 的序列,定义 SSM 隐状态为耦合双轨表示:
- • 单表示(Single Representation):捕获每个残基或配体原子的状态;
- • 对表示(Pair Representation):编码令牌对之间的几何交互。
蛋白质残基被聚合为残基级令牌,配体原子保持原子分辨率。这一设计在隐空间维持可控的计算量,同时以全原子坐标 作为可观测输入输出,保留了精细几何上下文。
3.2 几何编码器(Geometric Encoder )
编码器负责将当前全原子坐标 提取为强迫项(forcing term),类比 SSM 中 ,注入当前帧的几何信息:
编码器由两部分组成:
对几何特征(Pairwise Geometric Features):对每对令牌 计算不变几何特征——以 (甘氨酸用 )为蛋白质残基中心,以原子本身为配体中心,计算距离直方图和局部坐标系下的单位向量,拼接后通过 MLP 投影:
令牌级局部编码(Token-wise Local Encoding):采用 AtomAttentionEncoder(与 AlphaFold3 相同架构)处理全原子结构,汇总令牌内原子排布及其近邻化学环境:
3.3 状态转移模块(Transition Module )
这是 ATMOS 的核心传播器,负责将隐状态从 推进到 :
转移核的具体形式为:
其中 为广播的可学习时步嵌入, 为改进版 Pairformer 模块(4个 Pairformer block)。
Pairformer 的设计动机:标准 Pairformer 已具备单轨与对轨之间的信息交互能力,适合建模几何推理。ATMOS 在此基础上引入了双向信息流(Bidirectional Information Flow, BIF),在每个 block 开始时将单表示注入对表示:
z_{ij} ← z_{ij} + s_i + s_j # BIF:单 → 对(ATMOS 新增)
z_{ij} ← z_{ij} + TriangleMultiplication + TriangleAttention + Transition
s_i ← s_i + AttentionPairBias(s, z) + Transition这使得序列层面的信息能够更直接地反馈到对轨的几何推理,增强了动力学传播的质量。
架构超参数:单表示维度 384,对表示维度 128,三角乘法隐维度 128,三角注意力头数 4。
3.4 扩散解码器(Diffusion Decoder )
给定更新后的隐状态 ,解码器通过逆扩散过程生成下一帧坐标 ,采用 EDM 风格(Karras et al., 2022)的扩散参数化:
其中 , 为噪声扰动。解码器同时以 为条件,确保生成结构既满足化学合理性,又与轨迹历史动力学一致。解码器权重初始化自 Protenix(AlphaFold3 的开源复现),提供强力的几何先验。
解码采用随机微分方程(SDE),而非确定性 ODE——消融实验表明,SDE 的随机扰动对于再现生物分子构象的热力学多样性至关重要。
四、训练策略
4.1 Teacher Forcing 与子序列采样
为高效训练长轨迹数据,ATMOS 采用Teacher Forcing + 子轨迹窗口采样:从完整轨迹中采样起始索引 、时间步长 和窗口大小 ,以前 帧为上下文预测第 个目标帧,避免展开全轨迹的巨大内存开销。
4.2 损失函数
训练目标为两项损失的加权组合:
重建损失 (扩散目标):
其中 Align 通过 Kabsch 算法对齐,消除全局旋转/平移影响;权重 同时考虑原子实体类型(配体原子权重 11.0,蛋白质 1.0)和扩散时间步权重。
潜空间保真度损失 (距离图目标):
对潜变量中的对表示施加几何约束,通过预测 64-bin 距离分布的交叉熵损失,强制隐空间编码正确的空间关系。
超参数:,,。
五、推理:双层采样协议
5.1 设计动机
长程自回归生成中,误差会沿时间轴累积(drift),导致轨迹逐渐偏离物理合理区间。受生物物理学的多时间尺度运动理论(Henzler-Wildman & Kern, 2007)启发,ATMOS 引入分层的关键帧-插值策略。
5.2 关键帧生成器(Keyframe Generator )
以粗粒度时间步 ( 为上采样因子)训练,自回归生成稀疏锚定构象序列,捕获大尺度集体运动。
5.3 条件插值器(Conditioned Interpolator )
以细粒度时间步 训练,在相邻关键帧之间填充中间帧。插值器同时以起始帧 和终止关键帧 为条件,配合剩余时间 确保插值路径收敛于正确终点:
关键优势:相邻关键帧之间的所有中间帧可以并行生成(ATMOS-Parallel),在保持时序一致性的同时大幅提升采样效率。
5.4 训练规模
数据集 | 粗模型时间步 | 细模型时间步 | 上采样因子 |
|---|---|---|---|
mdCATH | 20 ns | 1 ns | 20 |
MISATO | 0.8 ns | 0.08 ns | 10 |
六、实验结果
6.1 数据集
mdCATH(蛋白质单体动力学):覆盖 5,398 个蛋白结构域,5 个温度条件,各 5 条模拟轨迹,以 1 ns 间隔采样。遵循 TEMPO 的数据划分:训练 1,000 个结构域,验证 50 个,测试 64 个;训练集与测试集平均序列相似度仅 18.93%,确保严格的泛化性评估。
MISATO(蛋白质-配体复合物动力学):300 K 下 8 ns 模拟,每条轨迹 100 帧。经预处理后:训练 12,786 个体系,验证 1,278 个,测试 100 个(随机采样)。
6.2 蛋白质单体动力学评估(mdCATH)
评估维度与指标:
维度 | 指标 | 说明 |
|---|---|---|
柔性预测 | Pairwise RMSD Pearson r | 构象集合整体多样性相关性 |
柔性预测 | Global RMSF r | 所有蛋白质残基 RMSF 的全局相关 |
柔性预测 | Per-target RMSF r | 每蛋白质内部 RMSF 相关(取中位数) |
分布精度 | Root mean W₂-dist | 原子位置分布的 Wasserstein 距离 |
分布精度 | MD/Joint PCA W₂-dist | 主成分空间中的分布相似度 |
分布精度 | % PC-sim > 0.5 | 主运动模式捕获率 |
集成可观测量 | Weak/Transient contacts J | 接触动力学的 Jaccard 相似度 |
轨迹精度 | t-RMSD Error | 构象变化幅度的时序误差 |
主要结果:
模型 | Pairwise RMSD r ↑ | Global RMSF r ↑ | Root mean W₂-dist ↓ | t-RMSD Error ↓ |
|---|---|---|---|---|
AlphaFlow-MD | 0.61 | 0.57 | 3.40 | — |
ESMFlow-MD | 0.46 | 0.47 | 5.00 | — |
MDGen | 0.65 | 0.67 | 3.86 | 2.71 |
TEMPO | 0.73 | 0.61 | 4.98 | 2.42 |
ConfRover | 0.69 | 0.62 | 4.52 | 2.50 |
ATMOS(本文) | 0.92 | 0.90 | 2.70 | 1.79 |
MD 参考 | 0.96 | 0.95 | 1.89 | 0.00 |
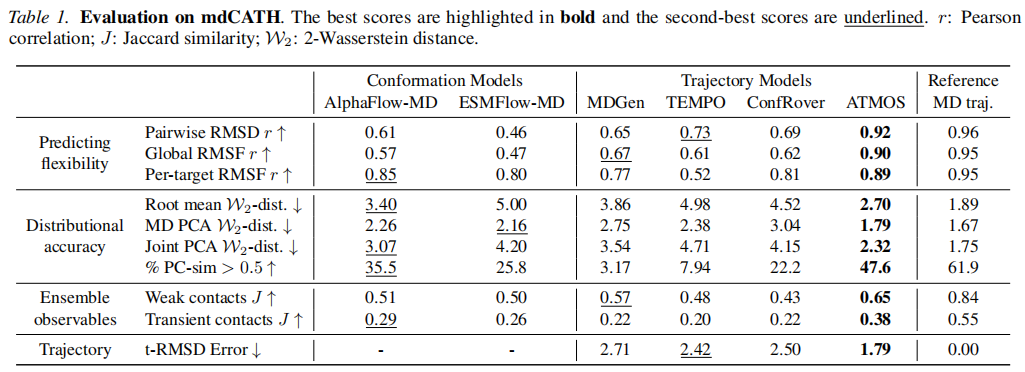
ATMOS 在全部指标上实现 SOTA,且各指标均接近 MD 参考轨迹水平。Pairwise RMSD 相关系数由最优基线(TEMPO,0.73)提升至 0.92,主成分捕获率(% PC-sim > 0.5)达 47.6%,远优于次优基线 AlphaFlow(35.5%)。
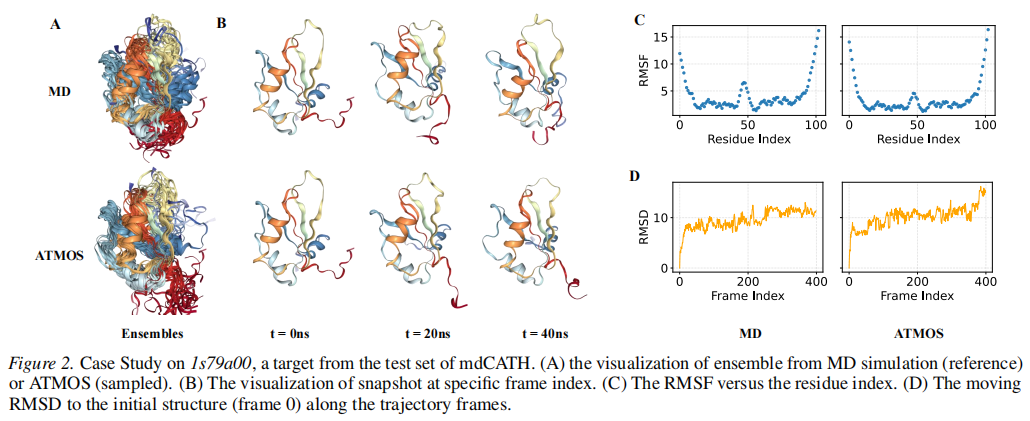
6.3 蛋白质-配体体系评估(MISATO)
方法 | per-target RMSF r ↑ | Root mean W₂-dist ↓ | Steric Clashes ↓ | t-RMSD Error ↓ |
|---|---|---|---|---|
NeuralMD-SDE | 0.574 | 0.698 | 7.667 | 0.698 |
DynamicBind | 0.678 | 1.470 | 7.113 | — |
Boltz-2† | 0.613 | 1.237 | 0.210 | — |
ATMOS(配体评估) | 0.746 | 0.670 | 0.030 | 0.480 |
DynamicBind(全系统) | 0.193 | 2.288 | 52.74 | — |
Boltz-2†(全系统) | 0.534 | 1.992 | 0.020 | — |
ATMOS(全系统) | 0.523 | 1.567 | 0.051 | 0.600 |
†注:Boltz-2 在完整 MISATO 数据集上训练,存在测试集数据泄露风险,且不生成轨迹,无法报告 t-RMSD Error。
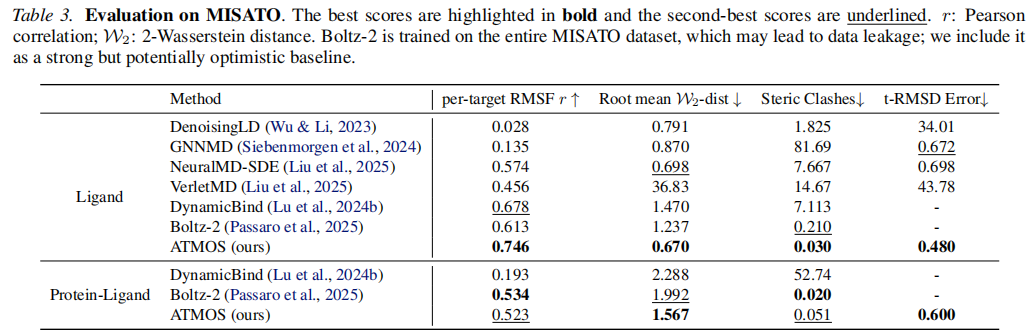
ATMOS 在配体动力学评估中四项全能最优,尤其在空间冲突(Steric Clashes,0.030)和时序精度(t-RMSD Error,0.480)上大幅领先,证明其生成的配体运动既物理合理,又具备时序一致性。
6.4 推理效率对比
以含 139 个氨基酸的蛋白质 1gnla01 为基准,生成 400 帧、400 ns 轨迹(RTX 4090 GPU):
方法 | 推理时间(小时) | 显存峰值(GB) |
|---|---|---|
MD 模拟(Amber24) | > 10 | < 1 |
ConfRover | 0.467 | 22.53 |
ATMOS | 0.174 | 3.445 |
ATMOS-Parallel | 0.044 | 6.117 |
ATMOS 相比 MD 快 > 60 倍,相比 ConfRover 快 2.7 倍,仅需 ConfRover 15% 的显存。并行化插值(ATMOS-Parallel)进一步将时间压缩至 0.044 小时,比 MD 快约 230 倍。
6.5 消融实验
配置 | Pairwise RMSD r | Global RMSF r | Root mean W₂-dist | t-RMSD Error |
|---|---|---|---|---|
ATMOS(完整) | 0.92 | 0.90 | 2.70 | 1.79 |
w/o 双层采样 | 0.88 | 0.84 | 3.06 | 2.03 |
ODE 替换 SDE | 0.73 | 0.64 | 3.89 | 3.22 |
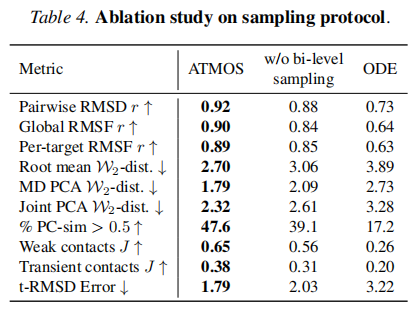
两项消融均验证了各组件的必要性:去除双层采样导致误差累积效应;将 SDE 替换为确定性 ODE 则大幅损失构象多样性,印证了随机性对生物分子热力学采样的本质重要性。
七、技术创新点总结
7.1 SSM 用于分子轨迹生成(首次)
SSM 此前主要用于语言和信号处理(S4、Mamba 等)。ATMOS 首次将 SSM 引入分子轨迹生成,赋予其与 MD 积分器类比的物理解释——递归隐状态对应相空间,线性推理复杂度打破了长序列建模的效率瓶颈。
7.2 全原子精度建模
相较于仅使用 或骨架帧(backbone frame)的粗粒化方法,ATMOS 在编码和解码阶段均操作全原子坐标,保留了侧链运动、配体构象等精细几何信息。
7.3 统一多分子体系框架
通过令牌化设计和分子身份标记,ATMOS 统一处理蛋白质单体和蛋白质-配体复合物,无需针对不同体系设计独立架构。
7.4 双层分层采样
关键帧-插值的两阶段策略实现了宏观构象变化与局部热涨落的解耦,并支持细粒度帧的并行生成,同时兼顾生成质量与推理速度。
7.5 预训练权重初始化
借助 Protenix(AlphaFold3 开源复现)的预训练参数初始化上下文嵌入层和扩散解码器,有效缓解了轨迹数据规模有限的问题,实现数据高效学习。
八、局限性与未来方向
8.1 当前局限
- • 双模型设置:关键帧生成器和插值器目前为独立训练的两个模型,增加了部署复杂度和资源需求;统一单模型尚未实现;
- • 固定时间步长:训练中采用固定时间步长 ,跨时间尺度的动力学(如多时间分辨率联合学习)尚未探索;
- • 数据集局限:mdCATH 训练集仅 1,000 个结构域,规模仍有限;MISATO 的蛋白质-配体体系仅覆盖部分化学空间;
- • 与 Boltz-2 的差距:在全系统 RMSF 相关性指标上略逊于 Boltz-2(0.523 vs 0.534),尽管后者存在数据泄露;
- • 稀有事件采样:对于能量垒极高的构象转变,模型是否能有效捕获尚未系统验证。
8.2 未来方向
- • 统一关键帧生成与插值为单一多尺度模型;
- • 随机化时间步长 ,使模型具备跨时间分辨率的泛化能力;
- • 扩展至核酸、蛋白质-蛋白质复合物等更广泛的生物分子体系;
- • 结合增强采样技术改善稀有构象转变的覆盖;
- • 以 ATMOS 为基础构建动力学基础模型(Dynamics Foundation Model),打通从静态结构预测到动力学模拟的完整链路。
九、对领域的意义
ATMOS 代表了计算生物学两条技术路线的深度融合:将 AlphaFold 路线(静态结构预测)中的强大几何建模能力,与深度序列模型(SSM)的高效时序推理相结合,并辅以扩散模型的高质量生成。
从更宏观的视角来看,该工作开辟了走向动力学基础模型的路径——若未来模型能够在更大规模的 MD 数据上完成预训练,则有望成为生物分子动力学研究的通用计算基础设施,对蛋白质工程、变构调节机制解析和基于结构的药物设计(尤其是考虑诱导契合效应的虚拟筛选)产生深远影响。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号