免疫组库基础分析14:基于NAIR包的TCR/BCR免疫组库公共簇分析

免疫组库基础分析14:基于NAIR包的TCR/BCR免疫组库公共簇分析

三兔测序学社
发布于 2026-04-17 16:49:55
发布于 2026-04-17 16:49:55
摘要:适应性免疫受体库测序(AIRR-Seq)是解析免疫应答、疾病标志物筛选的核心技术,TCR/BCR簇的跨样本共享性是关键研究切入点。NAIR(Network Analysis of Immune Repertoire)是基于R语言的免疫组库网络分析工具包,可高效实现公共TCR/BCR簇挖掘。
关键词:NAIR包;TCR/BCR;免疫组库;公共簇;网络分析
一、引言
T细胞受体(TCR)与B细胞受体(BCR)是适应性免疫的核心分子,其CDR3区序列多样性决定了抗原识别特异性。
在群体水平,公共簇指跨多个体/时间点共享、CDR3氨基酸序列差异≤1的相似克隆集合,反映保守免疫应答特征;
传统免疫组库分析侧重序列丰度与多样性,难以捕捉克隆间的相似性网络。
NAIR包基于图论与网络分析,将克隆视为节点、序列相似性视为边,通过聚类算法挖掘功能相关的受体簇,解决了多样本整合分析与表型关联挖掘的痛点。
二、环境配置与数据准备
2.1 包安装与加载
NAIR包托管于GitHub,需通过devtools安装,依赖igraph、tidyverse等基础包
# 安装NAIR包
install.packages("NAIR")
#
#第二种方式下载
devtools::install_github(
"mlizhangx/Network-Analysis-for-Repertoire-Sequencing-",
dependencies = TRUE,
build_vignettes = TRUE
)
# 加载包
library(NAIR)
set.seed(42) # 固定随机数,保证结果可复现2.2 模拟测试数据为避免真实数据限制,本文使用NAIR内置的simulateToyData()函数生成模拟AIRR-Seq数据,包含30个样本、每个样本30条序列,部分序列设计为跨样本共享,适配公共簇分析;后续关联簇分析将数据分为对照组与处理组,模拟表型关联数据。
2.2.1 公共簇分析模拟数
数据输出包含CloneSeq(CDR3序列)、CloneCount(克隆计数)、CloneFrequency(克隆频率)、SampleID(样本ID)四列,符合AIRR-Seq标准格式。
set.seed(42)
library(NAIR)
data_dir <- tempdir()
dir_input_samples <- file.path(data_dir, "input_samples")
dir.create(dir_input_samples, showWarnings = FALSE)
samples <- 30
sample_size <- 30 # (seqs per sample)
base_seqs <- c(
"CASSIEGQLSTDTQYF", "CASSEEGQLSTDTQYF", "CASSSVETQYF",
"CASSPEGQLSTDTQYF", "RASSLAGNTEAFF", "CASSHRGTDTQYF", "CASDAGVFQPQHF",
"CASSLTSGYNEQFF", "CASSETGYNEQFF", "CASSLTGGNEQFF", "CASSYLTGYNEQFF",
"CASSLTGNEQFF", "CASSLNGYNEQFF", "CASSFPWDGYGYTF", "CASTLARQGGELFF",
"CASTLSRQGGELFF", "CSVELLPTGPLETSYNEQFF", "CSVELLPTGPSETSYNEQFF",
"CVELLPTGPSETSYNEQFF", "CASLAGGRTQETQYF", "CASRLAGGRTQETQYF",
"CASSLAGGRTETQYF", "CASSLAGGRTQETQYF", "CASSRLAGGRTQETQYF",
"CASQYGGGNQPQHF", "CASSLGGGNQPQHF", "CASSNGGGNQPQHF", "CASSYGGGGNQPQHF",
"CASSYGGGQPQHF", "CASSYKGGNQPQHF", "CASSYTGGGNQPQHF",
"CAWSSQETQYF", "CASSSPETQYF", "CASSGAYEQYF", "CSVDLGKGNNEQFF")
# relative generation probabilities
pgen <- cbind(
stats::toeplitz(0.6^(0:(sample_size - 1))),
matrix(1, nrow = samples, ncol = length(base_seqs) - samples))
simulateToyData(
samples = samples,
sample_size = sample_size,
prefix_length = 1,
prefix_chars = c("", ""),
prefix_probs = cbind(rep(1, samples), rep(0, samples)),
affixes = base_seqs,
affix_probs = pgen,
num_edits = 0,
output_dir = dir_input_samples,
no_return = TRUE
)
# View first few rows of data for sample 1
head(readRDS(file.path(dir_input_samples, "Sample1.rds")))
#> CloneSeq CloneFrequency CloneCount SampleID
#> 1 CASSIEGQLSTDTQYF 0.02606559 2832 Sample1
#> 2 CASSEEGQLSTDTQYF 0.03718396 4040 Sample1
#> 3 CASSSPETQYF 0.03182726 3458 Sample1
#> 4 CASSIEGQLSTDTQYF 0.04615781 5015 Sample1
#> 5 CAWSSQETQYF 0.06006498 6526 Sample1
#> 6 CASSEEGQLSTDTQYF 0.03363123 3654 Sample1三、公共TCR/BCR簇分析全流程公共簇分析核心是先单样本筛选显著簇,再整合构建全局网络,分为3个核心步骤:单样本簇筛选、全局公共簇网络构建、下游可视化与分析。
3.1 步骤1:单样本显著簇筛选(findPublicClusters)
该函数为每个样本构建免疫组库网络,按节点数、克隆数过滤显著簇,输出元数据用于后续分析。
3.1.1 核心参数说明
input_files:样本文件路径向量input_type:输入文件格式(rds/rda/csv/tsv)seq_col:序列列名(本文为CloneSeq)count_col:克隆计数列名(本文为CloneCount)min_node_count:簇最小节点数(默认10)min_clone_count:簇最小总克隆数(默认100)output_dir:输出目录- The min_seq_length:序列最小长度
- drop_matches :排除字符类型
3.1.2 可运行代码
# 创建输出目录
dir_filtered_samples <- file.path(data_dir, "filtered_samples")
# 单样本簇筛选
findPublicClusters(input_files,
input_type = "rds",
seq_col = "CloneSeq",
count_col = "CloneCount",
min_seq_length = NULL,
drop_matches = NULL,
top_n_clusters = 3,
#默认top
20
min_node_count = 5,
min_clone_count = 15000,
output_dir = dir_filtered_samples
)
# 查看输出文件
list.files(dir_filtered_samples)
#> [1] "cluster_meta_data" "node_meta_data"
# 节点元数据(步骤2输入文件)
dir_filtered_node <- file.path(dir_filtered_samples, "node_meta_data")
head(list.files(dir_filtered_node))输出两个子目录:cluster_meta_data(簇水平元数据)、node_meta_data(节点水平元数据),仅需node_meta_data用于全局网络构建。3.2 步骤2:全局公共簇网络构建(buildPublicClusterNetwork)
合并所有样本的显著簇,构建全局网络,重新聚类得到公共簇,同时生成可视化图像。
3.2.1 核心参数说明
file_list:步骤1输出的节点元数据文件路径seq_col/count_col:与步骤1保持一致dist_type:序列距离算法(默认hamming,汉明距离)dist_cutoff:距离阈值(默认1,CDR3差异≤1)print_plots:是否打印网络图像
3.2.2 可运行代码
# 获取节点元数据文件路径
dir_filtered_samples_node <- file.path(dir_filtered_samples, "node_meta_data")
# Vector of file paths to node metadata from step 1
files_filtered_samples_node <-
list.files(dir_filtered_samples_node, full.names = TRUE)
#>files_filtered_samples_node 文件向量结果如下(部分文件展示)
#>[1] "F:/NAIR/findpublicclusters/filtered_samples/node_meta_data/1_Sample1.rds"
#>[2] "F:/NAIR/findpublicclusters/filtered_samples/node_meta_data/10_Sample10.rds"
#>[3] "F:/NAIR/findpublicclusters/filtered_samples/node_meta_data/11_Sample11.rds
# 构建全局公共簇网络
public_clusters <- buildPublicClusterNetwork(
files_filtered_samples_node,
seq_col = "CloneSeq",
count_col = "CloneCount",
size_nodes_by = 1,
print_plots = TRUE
)
3.3 步骤3:结果解析与下游分析
3.3.1 输出结果结构
# 查看输出对象结构
names(public_clusters)
# 节点元数据(每个节点对应一个克隆)
node_data <- public_clusters$node_data
head(node_data[, c("CloneSeq", "SampleID", "ClusterIDInSample", "ClusterIDPublic")])
# 簇元数据(每个簇的网络属性)
cluster_data <- public_clusters$cluster_data head(cluster_data[, 1:6]ClusterIDInSample:单样本内部簇ID
ClusterIDPublic:全局公共簇ID
网络属性:节点度、中介中心性、聚类系数等
3.3.2 簇标注与单簇可视化
# 标注全局最大6个簇的ID
public_clusters <- labelClusters(
public_clusters,
cluster_id_col = "ClusterIDPublic",
top_n_clusters = 6,
size = 7
)
# 查看标注后的图像
public_clusters$plots[[1]]
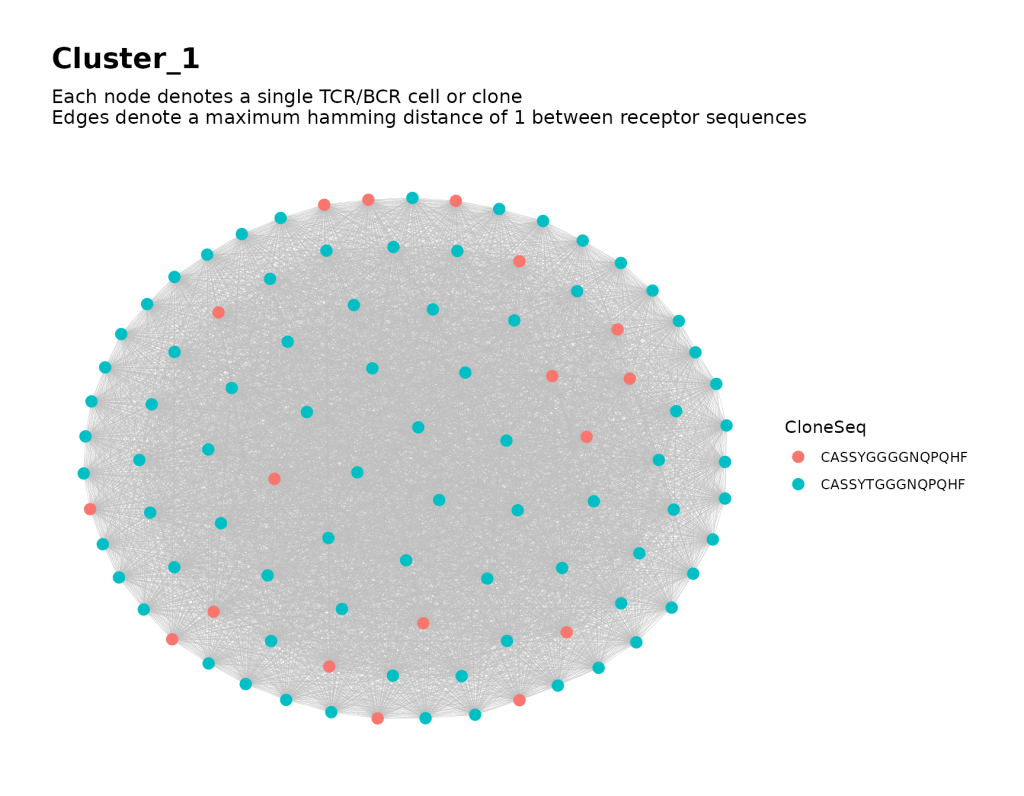
# 单独分析1号公共簇
buildNet(
public_clusters$node_data[public_clusters$node_data$ClusterIDPublic == 1, ],
seq_col = "CloneSeq",
color_nodes_by = "CloneSeq",
size_nodes_by = 3,
output_name = "Cluster 1",
print_plots = TRUE
)
四、核心参数调优与注意事项
4.1 序列距离算法选择
- 汉明距离(hamming):默认,适合氨基酸序列,计算速度快,要求序列长度一致。
- 莱文斯坦距离(lev):适合核苷酸序列,支持长度差异,计算成本高。
4.2 距离阈值设置
- 公共簇默认
dist_cutoff=1,即CDR3序列仅1个氨基酸差异,符合免疫组库共识。 - 研究保守序列可设为0(完全匹配),研究广谱相似簇可设为2。
4.3 聚类算法替换
默认使用igraph::cluster_fast_greedy(),可通过cluster_fun参数替换:
# 替换为Louvain算法
buildPublicClusterNetwork( file_list = files_filtered_node, seq_col = "CloneSeq", cluster_fun = igraph::cluster_louvain # Louvain聚类 )4.4 输入数据要求
- 每个样本独立存储为rds/csv等格式,列名统一。
- 必须包含序列列,克隆计数列为可选(用于丰度过滤)。
- 序列不含
*、_、|等非法字符,可通过drop_matches参数过滤。
参考文档:https://mlizhangx.github.io/Network-Analysis-for-Repertoire-Sequencing-/articles/public_clusters.html
免疫组库分析合集
【生信分析】免疫组库基础分析2-V/D/J基因使用频率可视化图
【生信分析】免疫组库基础分析3-基于V/D/J使用频率的聚类分析
【生信分析】免疫组库基础分析4-VJ/VDJ组合使用频率计算及可视化
【生信分析】免疫组库基础分析6-克隆子的分布分析(优势克隆与罕见克隆)
【生信分析】免疫组库基础分析10-CDR3 氨基酸理化性质分析
免疫组库分析11:Alakazam包【基因使用、多样性及氨基酸理化性质分析】使用说明
免疫组库分析12:TCR、BCR序列相似性网络分析-NAIR包介绍(1)
免疫组库基础分析13:TCR\BCR序列相似性网络分析_NAIR包介绍(2)
如果你觉得这篇博文对你有帮助,请点赞、收藏、转发!支持我们持续输出优质内容!
关注“三兔测序学社”,获取更多测序分析实用教程与前沿解读
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号