Science|Arc Institute: 蛋白质语言模型与上位效应引导的快速定向进化

Science|Arc Institute: 蛋白质语言模型与上位效应引导的快速定向进化

DrugOne
发布于 2026-03-25 16:22:17
发布于 2026-03-25 16:22:17
蛋白质工程受限于在高维序列空间中低效搜索协同突变组合。传统方法采用逐步叠加突变的方式,而机器学习方法则需要大量数据集或多轮实验,并受到成本高昂、长度受限的基因合成的瓶颈制约。
针对上述问题,Arc Institute研究人员于2026年2月19日在《Science》上发表文章,题为“Rapid directed evolution guided by protein language models and epistatic interactions”,其中斯坦福大学的生物化学教授Silvana Konermann、加州大学伯克利分校的生物工程教授Patrick D. Hsu为共同通讯作者。
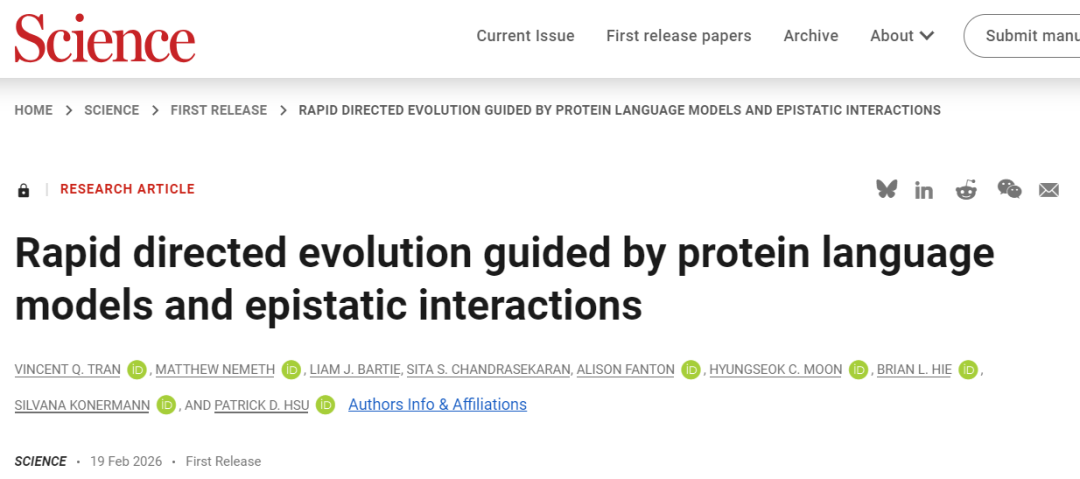
研究提出了一个能够系统性改造多突变体的快速进化框架MULTI-evolve。该方法结合了蛋白质语言模型或现有功能数据与上位效应建模,以预测协同突变组合。将MULTI-evolve应用于三种蛋白质,仅通过单轮机器学习引导的定向进化就实现了高达10倍的性能提升。MULTI-evolve为多种蛋白质类型和功能的端到端多突变体工程提供了一种精简高效的方法。
MULTI-evolve代码仓库:
https://github.com/VincentQTran/MULTI-evolve
背景
蛋白质驱动着所有生命形式的各种功能,通过定向进化和蛋白质工程,这些功能已催生了从工业用酶到人类治疗药物等众多应用。定向进化方法通过以加速的方式模拟缓慢的进化过程改进了蛋白质工程。近期,定向进化方法也借助机器学习得到了增强。通过利用模型进行计算机模拟变体筛选,机器学习引导的定向进化(MLDE)覆盖了更广阔的搜索空间,并减少了实验筛选的工作量。然而,这些模型的预测能力取决于学习目标蛋白中的上位效应,即一个氨基酸突变的功能影响可能会改变其他突变。没有学习这些上位效应会导致对增强型变体的预测不佳,特别是当训练数据集中功能增强型突变的比例较低时。此外,计算提名的变体很难用常规方法实际生成以供实验评估,尤其对于大蛋白而言。总的来说,当前方法不具备普适性,其应用效率低下,且通常依赖于针对特定蛋白质的机制性知识。
结果
MULTI-evolve框架的开发与基准测试
蛋白质工程需要在浩瀚的序列空间中导航,以找到能够增强功能的协同突变。传统的定向进化方法依赖于探索性极小的逐步行走,一次只评估一个新突变(图1A)。最近的MLDE方法通过计算机模拟变体筛选来进行更广泛的搜索;然而,准确的模型引导外推以得到增强型多突变体,依赖于多轮迭代的广泛实验验证(图1B)。为了克服这些限制,研究团队开发了MULTI-evolve,一个旨在单轮MLDE中直接"跳跃"到超活跃多突变体的框架(图1C)。该完整框架整合了三个关键组件,包括基于蛋白质语言模型(PLM)的突变发现、用于上位效应建模的机器学习以及高效的多点诱变。
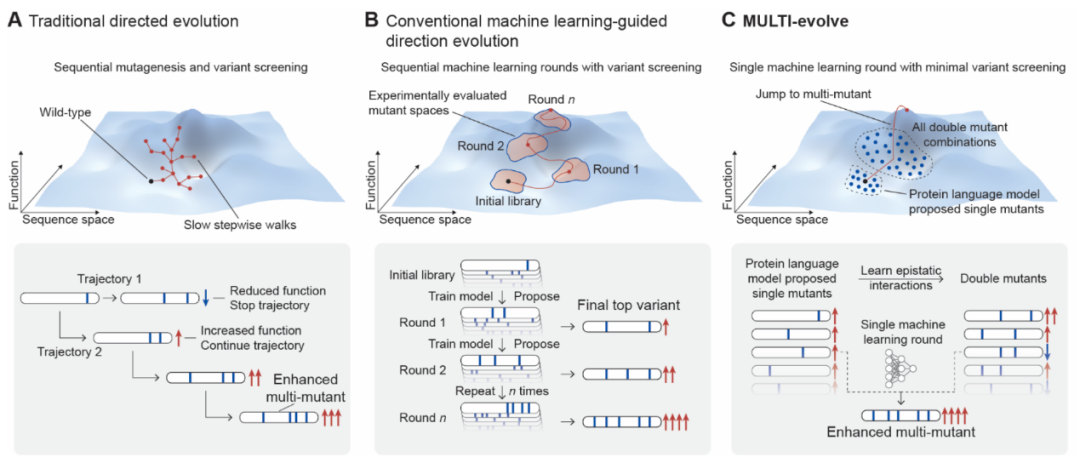
图1 使用MULTI-evolve工程化超活跃多突变体蛋白
虽然单个突变可以改善蛋白质功能,但功能的显著增强通常需要多个有益突变协同作用。结合多种PLM零样本方法将发现更多可供组合的有益突变(图2A),因而方法的集成可以减轻导致特定蛋白质失败的特定方法偏差。通过对73个不同的深度突变扫描数据集进行全面的基准测试,作者开发了一个PLM零样本集成方法,以全面识别功能增强型突变(图2B)。通过结合四种不同的PLM方法,该集成方法识别出超过20个功能增强型突变,而单独的PLM方法每种最多只能识别出11个功能增强型突变(图2C)。随后对跨越不同蛋白质家族的已发表深度突变扫描数据集上的FCNN性能进行了基准测试(图2D)。在所有测试的蛋白质中,与仅使用单突变体相比,在训练集中加入双突变体显著提高了预测性能,而加入三突变体则提供了额外的提升(图2E)。此外,在双突变体上训练的模型能够很好地外推至包含8-12个突变的组合(图2F)。总之,该基准测试结果表明双突变体实现了数据高效的训练,并能对跨不同蛋白质家族的多突变体进行强力的外推。

图2 MULTI-evolve框架的开发与设计
接下来,将MULTI-evolve模型与现有预测多突变体适应性的方法进行了验证比较(图2G)。MULTI-evolve模型在识别包含3到10个突变的高适应性多突变体的预测准确度和精度上均优于所有测试方法(图2H,I)。之后在先前建立的多点诱变方法基础上,通过优化反应条件对MULTI-assembly进行了系统性优化,以实现高构建效率并简化反应条件,从而将基因构建的流程时间减少了50%(图2J)。

在实践中,MULTI-evolve的操作如图2K所示。具体而言,从任何感兴趣的蛋白质开始,首先使用PLM零样本集成或现有的功能数据识别功能增强型单突变体。然后,对前约15个突变的所有成对组合进行实验表征,生成一个紧凑但信息丰富的数据集,该数据集捕获了关键的上位相互作用。这些数据用于训练MULTI-evolve模型,该模型学习上位效应景观并外推以预测所有可能的高阶组合的活性。在提名多突变体时,实验测试观察到最多包含七个突变的变体功能有所增强。基于此观察,建议选择包含最多七个突变的多突变体,按预测活性排序,并通过MULTI-assembly快速合成排名靠前的候选者以进行实验验证。
工程化超活跃APEX多突变体
APEX具有快速、高动态范围的读出特性,并且存在已知变体可供比较,特别是APEX2。使用PLM零样本集成方法,提名并实验评估了跨越62个位置的82个突变。通过集成方法,在APEX中识别出16个功能增强型突变(图3B)。通过整合具有互补进化偏差的方法,该集成捕获了任何单一方法都会错过的有益突变,并增加了总命中数,命中定义为活性超过野生型1.4倍以上的突变(图3C)。除A134P突变外,共同识别出15个先前未描述的突变,这些突变使APEX活性提高了1.4至2.7倍,且这些突变是通过一种或多种PLM方法发现的(图3D)。大多数突变的表面定位表明它们的影响可能通过改善蛋白质稳定性来介导(图3E)。除A134P外,唯一靠近活性位点的突变是I185V。I185配位活性位点中的钠离子,推断缬氨酸替换可能降低局部疏水性以改善配位(图3F,G)。总体而言,在APEX上的结果验证了PLM零样本集成方法识别功能增强型突变(如超增强的A134P突变)的能力,为MULTI-evolve构建更高阶组合提供了丰富的突变集合。
应用MULTI-evolve将A134P变体与新识别的这些突变结合起来。所得变体显示出显著的改进,相对于APEX的倍数提升在193到256倍之间,相对于A134P突变体则在3.6到4.8倍之间(图3H)。每个多突变体的活性都高于它们各自组成的双突变体组合,高出5.5至8.7倍,表明MULTI-evolve成功提出了具有改进协同效应的多突变体。随后探究MULTI-evolve是否可用于在不含高功能增强性A134P突变的情况下,跨位点利用上位效应来产生协同多突变体。双突变体集合展示了从协同到拮抗的各种类型上位相互作用。使用相同的训练和测试设置来建模这些上位相互作用,为每种突变负载选择并评估了前三名变体。尽管活性仍比A134P突变体低约2倍,但MULTI-evolve变体相对于野生型APEX显示出11到13.5倍的倍数提升值(图3I)。MULTI-evolve的性能超过了最近的MLDE框架EVOLVEpro,后者在五轮后仅能将活性提高至1.9倍。EVOLVEpro得到的最佳突变是G50R,这也是通过PLM零样本集成方法提名的一个突变。
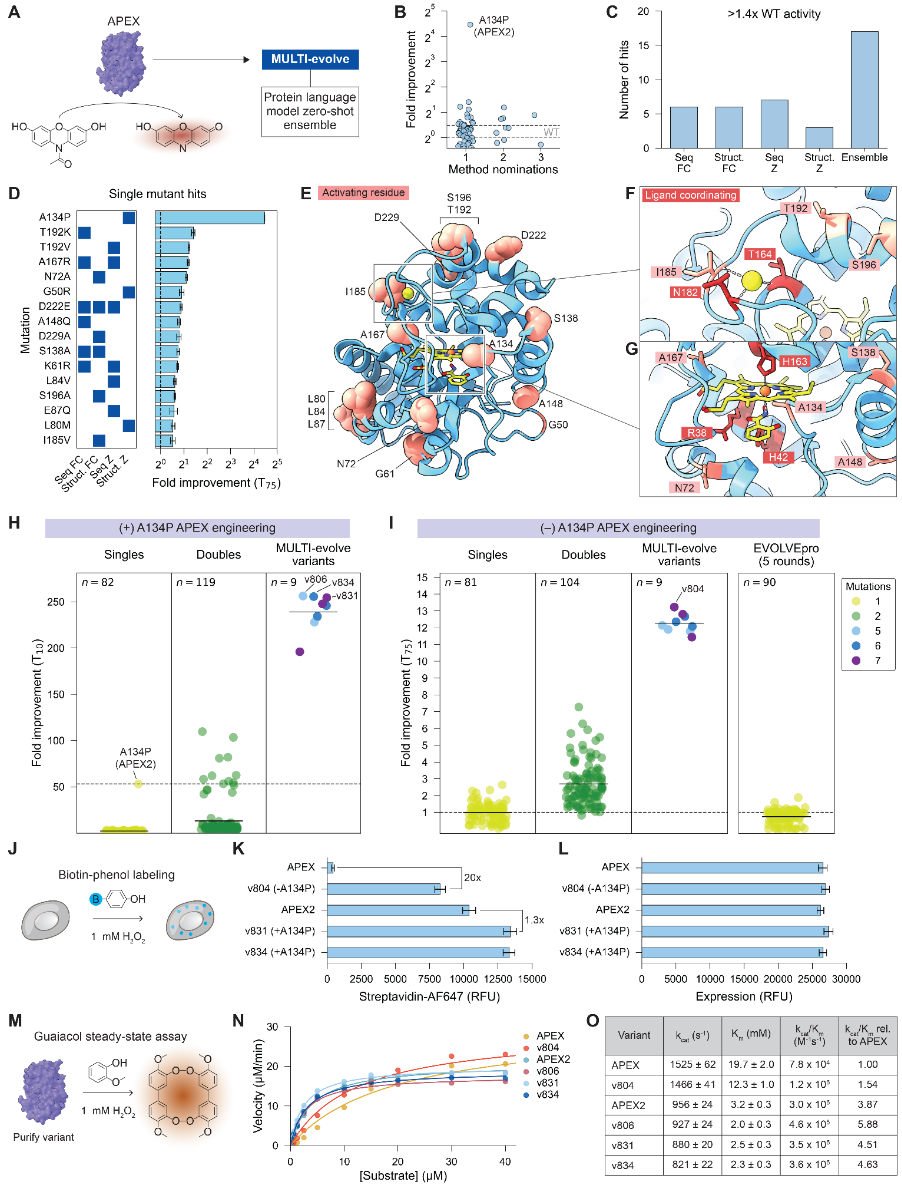
图3 使用MULTI-evolve工程化APEX
接下来,从机制上表征来自两项工程的最佳APEX变体。不含A134P的最佳变体v804,在生物素标记活性方面比APEX提高了20倍,达到了A134P突变体标记效率的80%(图3J,K)。含A134P的顶级变体v831和v834,与A134P突变体相比,生物素标记活性提高了1.3倍(图3K)。所有变体都保持了与APEX和A134P突变体相当的表达水平,证实活性改进并非由于蛋白质表达的改变(图3L)。与APEX相比,v804还显示出改进的Amplex Red氧化活性以及通过愈创木酚稳态测定法测量的酶动力学,其kcat/km提高了1.5倍(图3M至O)。对于含A134P的变体v806、v831和v834,与A134P突变体相比,也显示出增强的Amplex Red氧化活性和酶动力学,其kcat/km提高了1.2至1.5倍(图3N,O)。这些结果证实MULTI-evolve利用PLM提名的突变,识别了功能相关的上位相互作用,这些相互作用直接增强了生化催化作用,而非通过改善细胞表达来实现。
工程化超活跃dCasRx多突变体
为了展示该方法的普适性,接下来选择使用MULTI-evolve改造一个用于反式剪接应用的CRISPR-Cas蛋白系统(图4A)。几个突变显示出改进,范围从野生型的1.3倍到2.8倍(图4G)。结构分析显示增强突变聚集在功能区域。E282和S354P的多个突变发生在crRNA:目标RNA双链体附近,表明可能增强了目标RNA结合;P91K突变发生在crRNA直接重复环附近,可能通过突变为带更多正电荷的赖氨酸基团来改善crRNA识别(图4H)。MULTI-evolve变体显示出显著的改进,活性范围为野生型dCasRx的2.8倍到9.8倍(图4I)。当与靶向内源基因转录本的gTSM配对时,与野生型dCasRx相比,变体始终表现出增强的反式剪接活性,在所有基因上实现了平均3.9至4.5倍的改进(图4J至L)。在涉及gTSM和CIM两者的RESPLICE背景下,这些变体在所有基因上保持了一致的反式剪接效率增强,显示出平均1.3倍的改进(图4M至O)。与APEX类似,MULTI-evolve有效地模拟了dCasRx的上位效应景观,以发现高度增强的变体。此外,该dCasRx工程证明MULTI-evolve可以利用不同的突变来源进行工程改造,从计算性的PLM预测到实验筛选,无论可用的起始数据如何,都为其提供了灵活性。
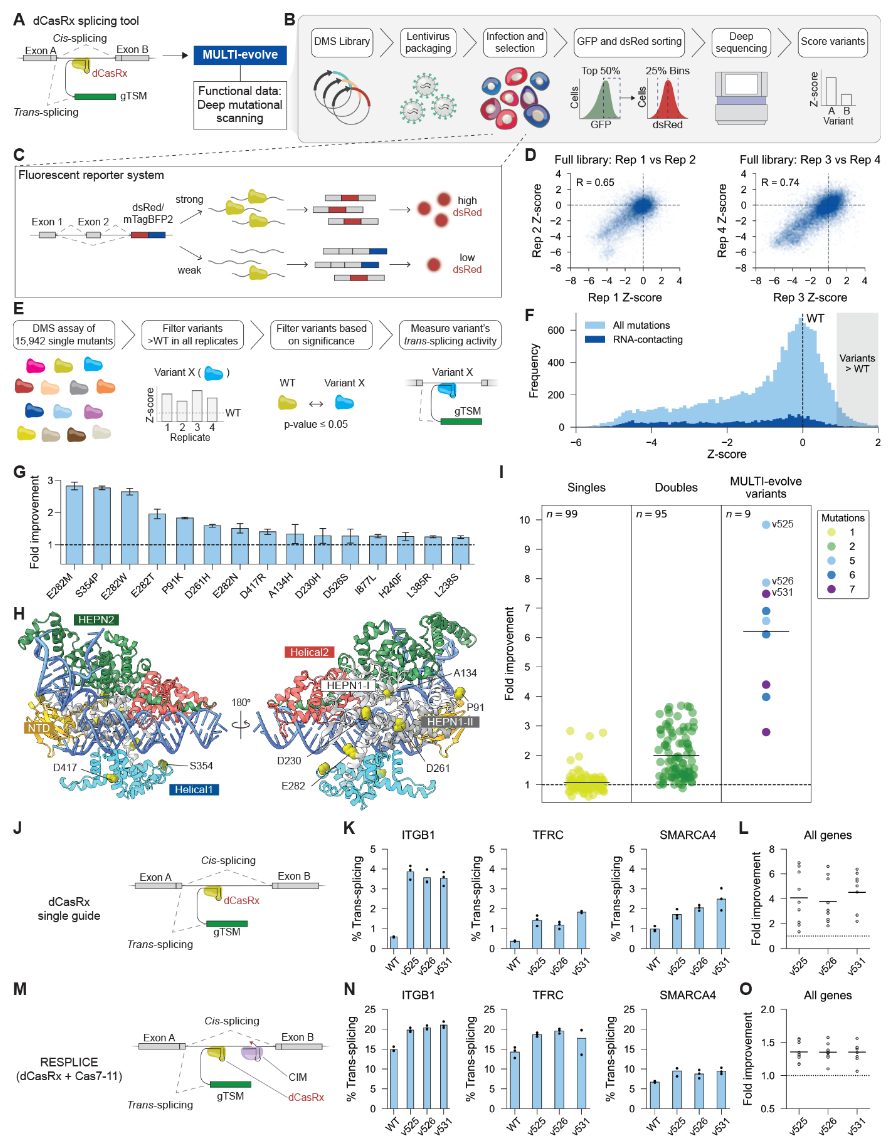
图4 使用MULTI-evolve工程化基于dCasRx的剪接工具
多目标抗体设计
为了展示MULTI-evolve在多目标优化中的适用性,作者选择了HuABC2,一种靶向CD122的抗体,该抗体与包括白癜风、乳糜泻和嗜酸性食管炎在内的自身免疫疾病相关(图5A)。图5结果突显了MULTI-evolve的关键优势:它能够学习功能增强型突变和特性特异性的上位效应架构,从而预测能跨竞争目标进行优化的多突变体。MULTI-evolve可以进一步将低纳摩尔结合剂的结合亲和力从2.7 nM提高到1.0 nM,同时提高表达产量。MULTI-evolve的模块化框架可以轻松扩展到任何可测量的蛋白质特性。通过整合针对血清半衰期、免疫原性或溶解度等额外参数的测定方法,相同的上位效应学习方法可以同时在更多维度上进行优化。该框架的数据高效训练特性使得并行表征多个读出的变体变得切实可行,从而能够从单次实验活动中实现全面的多参数优化。在此,同时改进了抗CD122抗体的表达量和结合亲和力,展示了MULTI-evolve在治疗性蛋白质开发中的能力,因为在此类开发中成功需要平衡可制造性、功效、安全性和药代动力学特性。
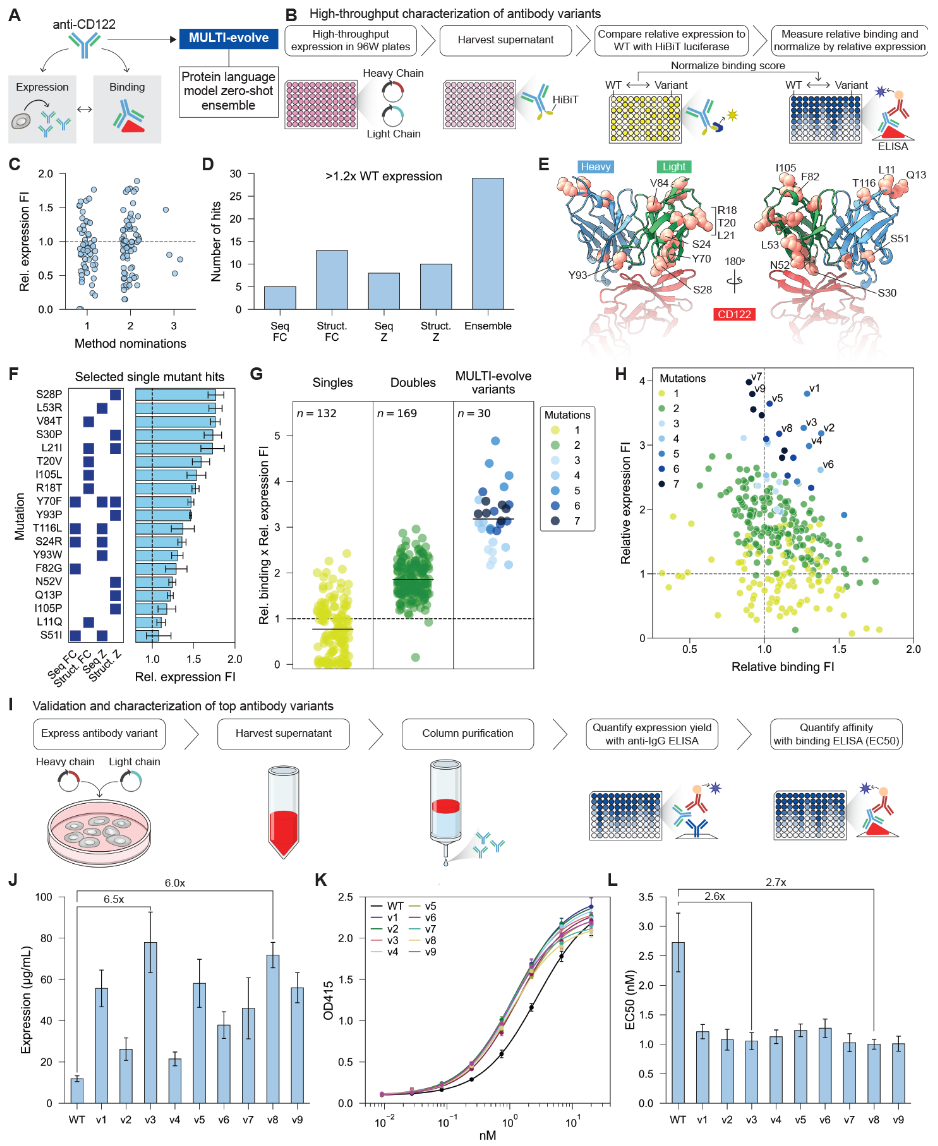
图5 使用MULTI-evolve进行多目标抗体设计
总结
本文提出了一个机器学习引导的框架MULTI-evolve,它通过将进化性PLM先验知识与上位效应建模相结合以获取协同的高阶组合,从而克服了蛋白质工程中巨大搜索空间的挑战。在三种不同的蛋白质APEX、Cas13d和抗CD122抗体上,改造出了性能提升2倍到256倍的变体,展示了广泛的适用性和多目标优化能力。无论初始突变是通过PLM计算识别,还是通过功能筛选实验识别,其预测多突变体活性的能力都是稳健的。通过MULTI-assembly实现了一个端到端的平台,该平台对复杂多突变体实现了40%-70%的合成效率,避免了主观的诱变寡核苷酸设计,并消除了商业DNA合成的瓶颈。最后,开发了一个MULTI-evolve设计工具,用于训练模型、预测多突变体以及设计用于克隆的MULTI-assembly寡核苷酸。总之,该工作建立了一个用户友好的框架,可以针对任何目标蛋白质快速设计复杂的多突变体。
参考链接:
https://doi.org/10.1126/science.aea1820
--------- End ---------
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号