文献分享--空间转录组学高分辨率绘制宿主-肠道微生物组生物地理分布图
原创
文献分享--空间转录组学高分辨率绘制宿主-肠道微生物组生物地理分布图
原创
追风少年i
发布于 2026-03-18 10:46:09
发布于 2026-03-18 10:46:09
作者,Evil Genius
生活总是坎坎坷坷,一点都不顺,不是这儿拌一下,就是那儿磕破了。
目前visium低精度发表的文章,大约1500篇左右,其中包括方法文;而visium HD,发表了大约30篇,而且我看了visium HD文章大约20篇,感觉还是和visium差异挺大的。
首先最明显的感觉就是,visium可以作为文章的主体部分,但是visium HD目前还没有这样的文章,很多分析的问题,visium HD还没有彻底的解决。
说起来分析分子niche、细胞niche,邻域通讯等内容,但是真正实施到visium HD上,仍然是困难重重;到目前为止我经手了十几个HD的项目,可以说客户的期待值是100,真正落地连20都没有,因为客户大多想以visium HD为主发表文章,实际却是摸着石头过河,全是坑。
现在风气已经不太好了,实验产品标准化交付,钱货两清,但是分析却无法做到这样,大部分客户都以为给了钱就能做到文章的水准,实际这才是科研的开始,需要不断的探索,试错,底层的东西,是无法标准化的,需要我们花费大量的心思来思考,来总结经验,所以为发文章最好的两个阶段,一是学生阶段,一门心思学习新的技能,探索,提升认知,发表高水平论文;二就是编制内阶段,无后顾之忧,有试错的空间,进行大量的总结,最后研究明白,所以说科研是一场马拉松,我们需要具有科研精神的人,需要长时间钻研课题的人,而这种人,据我的观察,只有编制 + 兴趣,才可能出现,我之前的生信分析的领导,大多数都不再做分析了,因为并不挣钱,例如周运来,现在转行去做市场销售了,只有转行才能还得起每月过万的房贷,从我的现状来看,放弃科研精神,估计也是必然的。
希望大家进入编制的过程中,科研的兴趣还能保持。
今天我们分享文献,visium + Stereo-seq + 微生物。

通过探索在空间RNA测序中利用酶促法对微生物RNA和宿主RNA进行原位加尾,来绘制微生物组和宿主图谱。
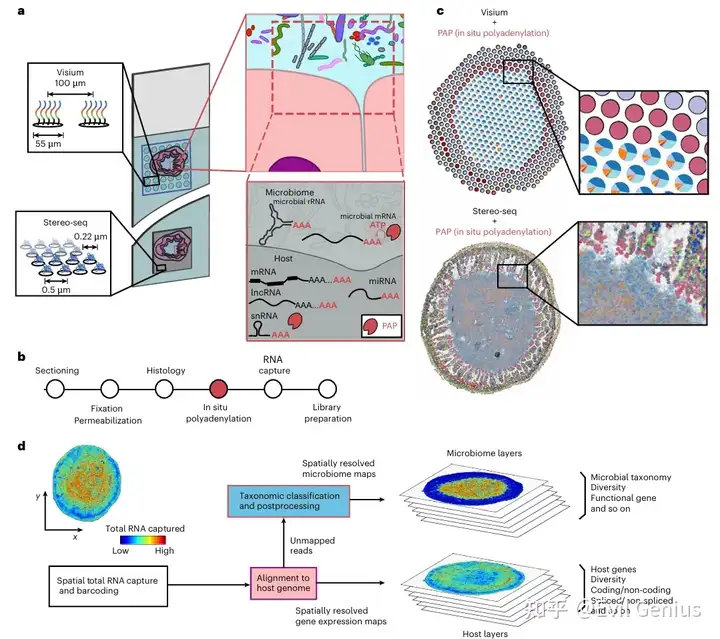
结果1、基于原位加尾技术的微生物组-宿主相互作用空间图谱构建
核心方法:
研究团队利用一种结直肠癌小鼠模型(APC缺陷),在其肠道四个不同区域(近端小肠、回肠、盲肠、结肠)采集新鲜冷冻组织样本。他们在Visium空间转录组学平台上,在标准流程之外,创新性地引入了酶促原位加尾步骤。该步骤旨在增强对微生物RNA(尤其是非聚腺苷酸化RNA)的捕获效率。为评估其效果,实验设置了未经原位加尾处理的对照组(近端组织切片)。通过测序,每个样本平均获得1.56亿条读取序列,每个捕获点约31,250条读取。
关键发现与验证:
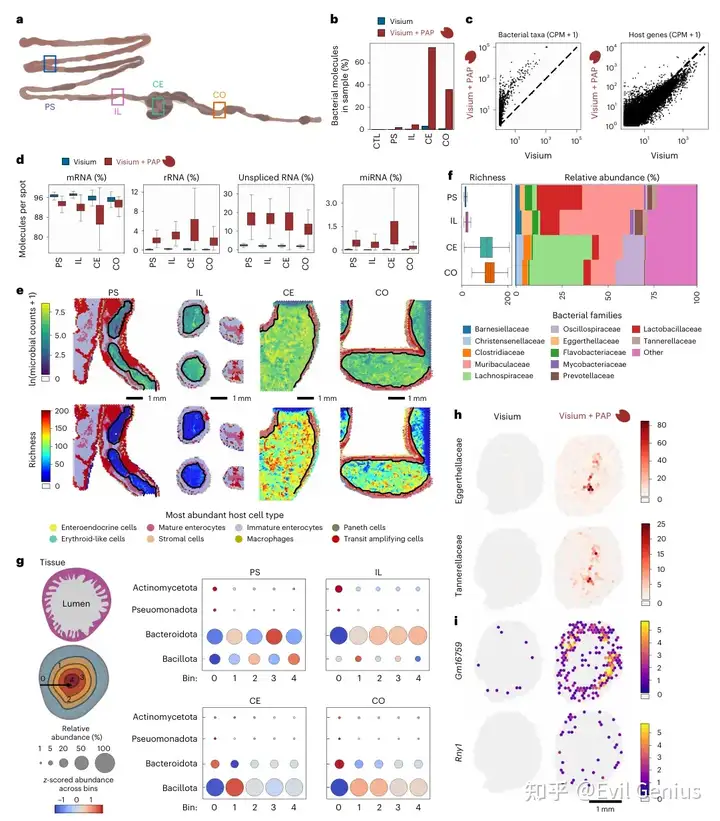
原位加尾显著增强微生物RNA捕获:
效率提升:与对照组相比,原位加尾技术使细菌RNA的捕获富集了高达99倍,尤其改善了对低丰度类群(如Tannerellaceae科、Eggerthellaceae科)的检测。
类群覆盖:对大多数细菌类群的捕获均有提升,同时保持了对宿主基因的高效捕获。病毒和古菌的RNA在近端小肠的捕获量也分别提高了10倍和6倍。
结果验证:将空间RNA-seq测得的微生物组成谱与批量宏转录组测序结果进行对比,在微生物生物量高的盲肠和结肠区域显示出高度一致性,验证了该方法的可靠性。在小肠区域一致性稍弱,这可能归因于该区域生物量低、易产生假阳性以及微生物在亚毫米尺度上的高度空间异质性。
原位加尾扩展了对宿主RNA的检测范围:
除了微生物RNA,该方法还显著改善了对多种类型宿主RNA的捕获,包括未剪接mRNA(可能代表新生转录本)、核糖体RNA(rRNA)、微小RNA(miRNA)、长链非编码RNA(lncRNA)等。这使得能够同时分析宿主细胞的动态响应和基因调控活动。
识别出在不同肠道区域具有特异性空间表达模式的非编码RNA,如回肠富集的lncRNA Gm16759(调控T细胞分化)、近端小肠的Gm31992以及盲肠和结肠的Gm56583和miR9-3hg(与人类癌症相关)。
揭示了肠道微生物组的空间组织模式:
纵向梯度(沿肠道长度):从近端小肠/回肠到盲肠/结肠,每个捕获点检测到的微生物分类单元(属水平)数量显著增加,从平均14.1个增至114.4个。不同细菌科(如乳杆菌科、毛螺菌科等)的丰度随区域变化,这与已知的肠道生态位分化相符。
横向梯度(从组织到肠腔):通过将组织切片按距肠腔距离分仓分析,发现不同细菌门(如放线菌门、拟杆菌门、芽孢杆菌门)在组织-黏液-肠腔轴线上呈现出偏好性分布。例如,在小肠,放线菌门和假单胞菌门更靠近组织/黏液层,而拟杆菌门则更趋向于远离组织的肠腔区域。在盲肠和结肠,芽孢杆菌门在所有区域占主导,但拟杆菌门在组织层富集。
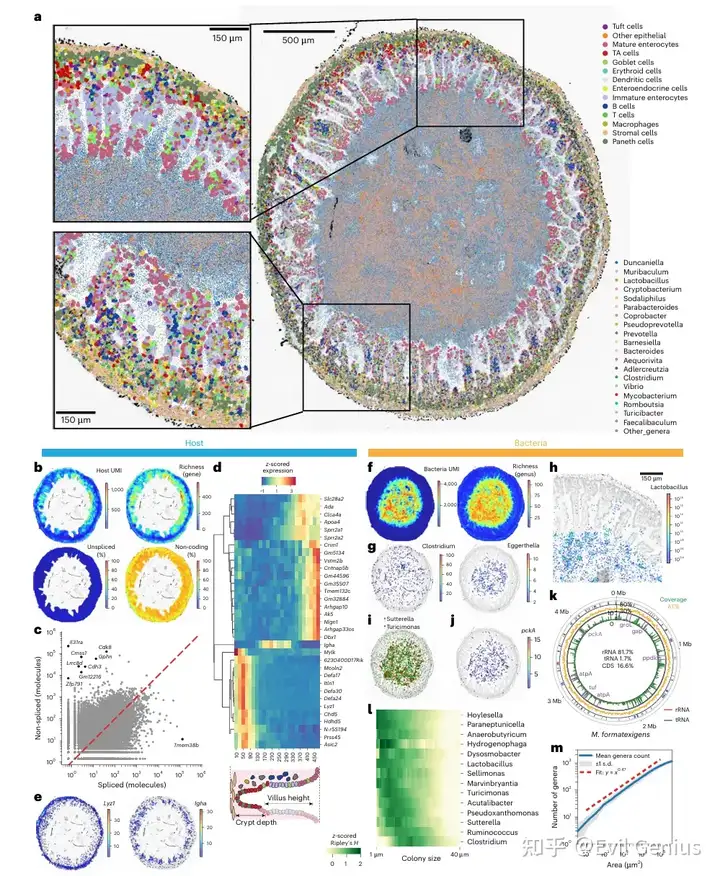
结果2、高分辨率下解析微生物组-宿主互作图谱
核心方法:
研究团队在Visium平台的基础上,进一步采用了分辨率高达~0.5微米/像素的Stereo-seq平台,并对Visium样本的相邻组织切片(小鼠回肠等)进行了分析。他们再次应用了原位加尾技术,以同时捕获宿主和微生物RNA,并结合成像数据与单细胞RNA测序数据,将宿主基因表达精确定位到单个细胞类型。
主要发现:
宿主基因表达的空间精细图谱:
编码基因:宿主基因表达呈现显著的空间非均匀性。肠绒毛顶端的成熟肠细胞显示出更高水平和更多样化的基因表达。
非编码与新生RNA:原位加尾技术再次验证了其对非编码RNA和未剪接mRNA(新生RNA)捕获的显著提升。未剪接mRNA(占宿主RNA的22.5%)在隐窝基部富集,可能与 transit amplifying 细胞的快速更新有关,其中包含与结直肠癌相关的癌基因(如Cdk8)。
区域特异性:非编码RNA(如6230400D17RiK)在靠近肠壁的区域高表达,而Ada基因则在绒毛顶端富集。
免疫相关基因:成功检测到参与微生物反应的基因空间分布,如潘氏细胞(隐窝基部)表达的溶菌酶Lyz1和防御素,以及固有层浆母细胞表达的IgA重链片段Igha。
微生物组的微尺度空间组织:
密度与多样性:观察到靠近宿主组织边界处细菌负荷和多样性较低。
空间分布异质性:不同菌属展现出截然不同的空间偏好。例如,梭菌属(Clostridium)分布相对均匀,克雷伯氏菌属(Klebsiella)富集于绒毛顶端,而埃格特菌属(Eggerthella)则远离组织边界。
菌落形成与大小:分析发现54个菌属显示出显著的菌落形成(空间自相关)。通过Ripley's H分析估算出不同菌属的菌落大小差异显著:乳杆菌属(Lactobacillus)菌落较小(半径<10微米),Turicimonas属菌落中等(~10微米),而梭菌属菌落较大(>30微米)。
菌属间互作:空间相关性分析揭示了不同菌属间的强关联,例如Turicimonas和Sutterella在空间上共定位。
沿肠道纵轴的微生物群结构变化:
高分辨率结果验证了低分辨率平台(Visium)的观察:从近端小肠到结肠,不同细菌门和科沿组织-肠腔轴线的分布模式具有区域特异性,并与已知的肠道生物地理学一致(如近端结肠中,毛螺菌科和颤螺菌科富集于组织边界,乳杆菌科和拟杆菌科则在肠腔更突出)。
菌落形成模式在不同肠道区域也有所不同:回肠和远端区域能观察到清晰的乳杆菌、Turicibacter和梭菌菌落,而近端小肠的菌落则更小。
细菌基因表达的检测:
通过将微生物读数比对到丰度最高的六个物种的基因组,成功检测到细菌的功能基因表达。例如,与快速生长相关的groL基因、与糖代谢相关的ppdk、pckA和gap基因,以及看家基因atpA,这些信号主要源自肠腔。不同肠道区域细菌RNA的组成(rRNA、tRNA、mRNA)也存在差异,为理解细菌在原位的生长和功能状态提供了线索。
物种-面积关系(SAR):
首次在肠道微生物组中量化了物种-面积关系。研究发现,观察到的菌属数量与取样面积在从16 µm² 到 0.16 mm²的三个数量级范围内遵循幂律分布,其幂指数(0.47)相对较高,表明小鼠回肠微生物组具有较高的空间分散性,这与动植物生态系统中的观察结果一致。
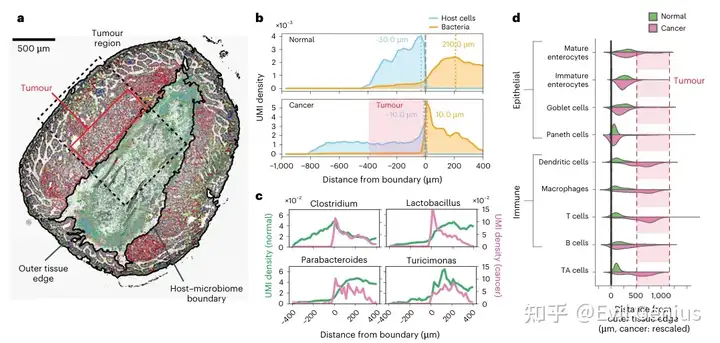
结果3、肿瘤相关微生物组空间重构
重点比较了肿瘤边缘区域与正常组织区域在宿主细胞和微生物空间组织上的差异。
微生物的空间分布发生剧变
正常组织:微生物的最大密度区域位于距离宿主绒毛100-200微米处,即微生物与组织之间存在一个无/低微生物的间隙。
肿瘤组织:微生物的最大密度区域直接位于肿瘤组织的边界处,微生物与肿瘤细胞紧密接触。
特定菌属向肿瘤边缘富集
丰度最高的梭菌属(Clostridium),以及乳杆菌属(Lactobacillus)和副拟杆菌属(Parabacteroides)在肿瘤边缘显著富集,与肿瘤组织紧密关联。而在正常组织中,这些菌属则远离组织边界。
乳杆菌属和Turicimonas在肿瘤区域依然表现出菌落形成的特征(半径约10-20微米)。
宿主细胞类型的空间重排
正常组织:最接近宿主-微生物边界的细胞是成熟肠上皮细胞,其后是未成熟肠上皮细胞。潘氏细胞位于隐窝基部,符合预期的组织结构。
肿瘤组织:肿瘤区域富集了肿瘤相关的 transit amplifying 细胞以及多种免疫细胞,包括树突状细胞、巨噬细胞和CD8+ T细胞。而负责产生黏液的杯状细胞则因肿瘤块的挤压而远离组织-微生物边界。
基因表达水平的变化
在肿瘤亚区域,部分基因(包括癌症相关基因 Pkm)的表达位置向肠腔方向移动,提示肿瘤可能改变了宿主细胞的基因表达程序及其空间定位。
观察结果的验证与延伸
相邻回肠切片的额外数据以及另一只独立小鼠的数据均验证了上述观察结果。此外,数据还提示肿瘤与微生物的接近程度可能与肿瘤大小存在相关性。
简单看一下方法

生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号