贝叶斯多模型推理提高系统生物学模型的确定性

贝叶斯多模型推理提高系统生物学模型的确定性

CreateAMind
发布于 2026-03-11 17:36:16
发布于 2026-03-11 17:36:16
Increasing certainty in systems biology models using Bayesian multimodel inference
贝叶斯多模型推理提高系统生物学模型的确定性
https://www.nature.com/articles/s41467-025-62415-4


数学模型对于研究细胞内信号网络的结构和行为至关重要。由于难以完整观测细胞内信号通路中的中间步骤,通常采用现象学近似方法来构建模型。因此,可以为同一条通路建立多个模型。这带来了模型选择的挑战,并降低了预测的确定性。本文中,我们探讨了贝叶斯多模型推断(MMI)作为一种提高系统生物学预测确定性的方法,这种方法在希望利用一组可能不完整的模型时尤为相关。通过使用现有的细胞外调节激酶(ERK)通路模型,我们展示了MMI能够成功整合多个模型,产生对模型集合变化和数据不确定性具有鲁棒性的预测结果。随后,我们利用MMI识别实验测量到的亚细胞位置特异性ERK活性的可能机制。本研究强调了MMI作为一种严谨方法,可提高对细胞内信号活动预测的确定性。
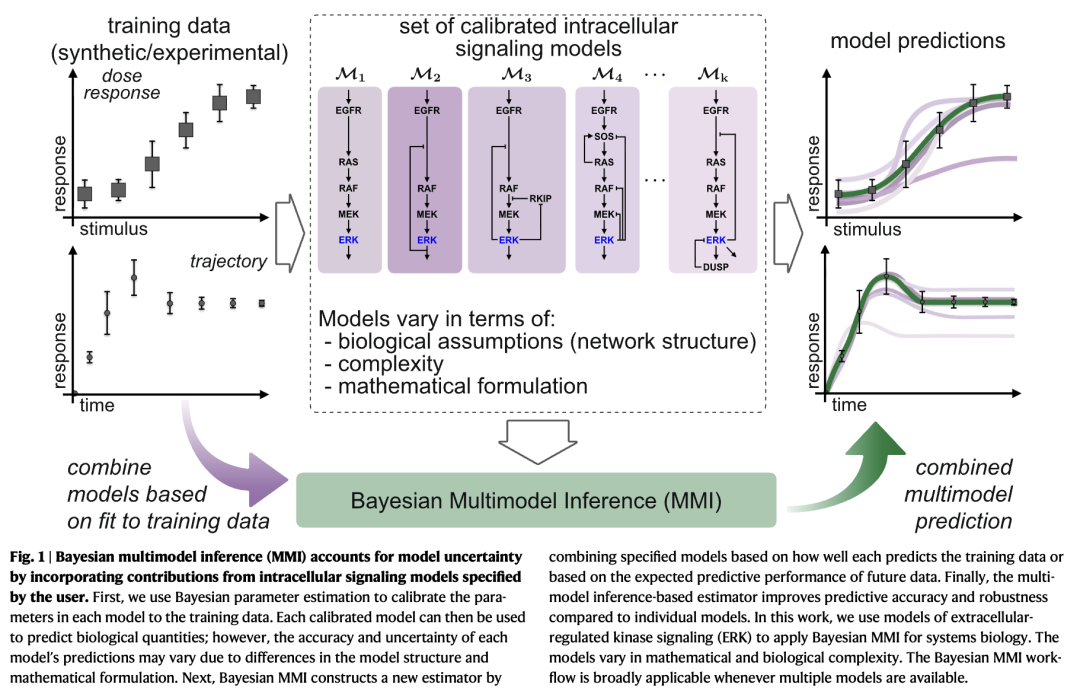
当前分子工具1,2 和高分辨率显微技术3,4 的发展推动了细胞内信号转导领域的发现,例如信号通路的空间调控。这些发现往往需要新的数学模型来提供机制性见解。此外,数学模型还能生成可实验验证的细胞内信号活动预测5–7。然而,系统生物学中的一个关键挑战是:当存在大量未知因素,且现有数据无法观测系统中所有组分时,如何构建模型。因此,针对同一条信号通路,可能产生多个在简化假设和模型结构上各不相同的数学模型。例如,在流行的BioModels数据库中搜索细胞外调节激酶(ERK)信号级联的模型,仅使用常微分方程的模型就超过125个8,9。尽管每个模型都是为特定实验现象而开发的,但若能综合考虑各种简化假设,则有助于提高预测的确定性和准确性。在本研究中,我们探讨两个重要问题:(1)如何量化模型结构不确定性(即模型不确定性)对模型预测的影响?(2)如何减少模型选择的不确定性,以提高细胞内信号预测的确定性?以ERK信号通路为模型系统,我们表明,贝叶斯多模型推断(MMI)通过利用现有数据并综合所有用户指定的模型,有助于应对模型不确定性。
系统生物学中的不确定性量化旨在理解模型假设、推断参数以及数据不确定性如何影响模型预测10,11。贝叶斯参数估计通过从训练数据中估计未知参数(如反应速率常数和平衡系数)的概率分布,定量评估参数不确定性10,12,13。然而,在模型层面,系统生物学中尚未常规采用明确处理模型不确定性的方法。当存在多个候选模型时,系统生物学通常采用信息准则(如赤池信息准则AIC)14,15 或贝叶斯因子16 进行模型选择,以挑选出单一“最优”模型14,17。基于单一模型得出的一组预测结果进行推理,远比综合多个模型的预测更容易。然而,由于现有的实验数据往往稀疏且带有噪声,这类方法可能因引入选择偏差和错误表征不确定性而限制预测性能14,18。
多模型推断(Multimodel inference)通过纳入所有指定模型的贡献,减少选择偏差并考虑模型不确定性13,14,18–21。理论研究表明,MMI能够通过降低不确定性、增强对建模假设的鲁棒性来提升预测性能14,18,22–24。MMI方法通过对所有提供的模型进行加权平均来整合模型预测23–25。权重选择方法包括参数共识估计23,24、基于频率学派的赤池加权法14、贝叶斯模型平均(BMA)19、伪贝叶斯模型平均(pseudo-BMA)18,22 以及预测密度堆叠法(stacking)22。
此前在系统生物学中应用MMI的研究主要局限于少数几种可用方法,尤其是基于信息准则和BMA的方法。Stumpf等人对生物网络推断中的MMI进行了理论分析,其中网络结构由数据推断得出20,21。基于这些分析以及对蛋白质-蛋白质相互作用数据的有限应用,作者得出结论:选择一组高质量的模型对于构建准确的MMI估计至关重要,即使真实网络不在候选模型集合中,MMI仍能改善对生物网络结构的预测。最近,Beik等人使用BMA结合MMI,从多个实验数据集中筛选出与肿瘤生长机制一致的候选模型26。然而,MMI尚未被用于提高系统生物学中细胞内信号预测的确定性。
在本研究中,我们扩展了贝叶斯系统生物学的工具集,将MMI作为一种强大方法来应对模型不确定性。我们以细胞外调节激酶信号通路(ERK)为例进行深入分析。具体而言,我们选取了十个强调ERK通路核心机制的模型,并利用Keyes等人27 的实验数据,通过贝叶斯推断估计其动力学参数。首先,我们证明了在多个不同的实验和合成数据集上,MMI能够提高模型预测的确定性。其次,我们发现基于MMI的预测对所提供ERK模型集合的组成变化以及数据不确定性的增加均具有鲁棒性。最后,我们应用MMI研究驱动ERK活性的机制,这些活性被Keyes等人27 观察到定位于特定亚细胞区域。我们将这种ERK活性称为“亚细胞位置特异性ERK活性”。细胞内信号的亚细胞定位被认为在区分细胞对异质刺激的不同响应中起重要作用,并可通过现代分子工具观测到27。我们的研究发现,贝叶斯MMI使我们能够比较关于何种机制驱动位置特异性ERK信号的不同假设,并表明:要准确捕捉所观察到的动力学特征,必须同时考虑Rap1激活和负反馈强度在不同亚细胞位置上的差异。我们得出结论:当存在多个同一信号通路的模型时,MMI通过一种系统化的方法同时处理模型不确定性和模型选择问题,能够有效提高预测的确定性。
结果 贝叶斯多模型推理

本研究中采用的贝叶斯多模型推理流程如图1所示:首先,我们通过贝叶斯推理估计未知模型参数,将现有模型校准至训练数据;然后,我们利用多模型推理方法将各模型得到的预测概率密度进行组合;最后,提供系统生物学研究中重要变量的改进型多模型预测。贝叶斯多模型推理通过取每个模型预测密度的线性组合来构建预测器。
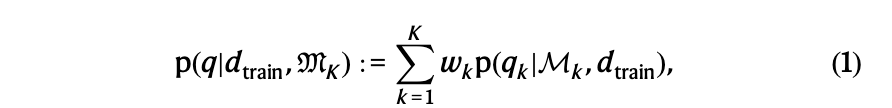

伪贝叶斯模型平均分配模型权重,基于未见数据的预期预测性能,通过预期对数点预测密度(ELPD)来衡量。ELPD通过计算预测和真实数据生成密度之间的距离来量化新数据的预期预测性能,并定义为:
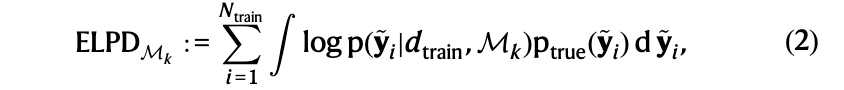

ERK 信号通路模型的差异体现了模型形式的不确定性
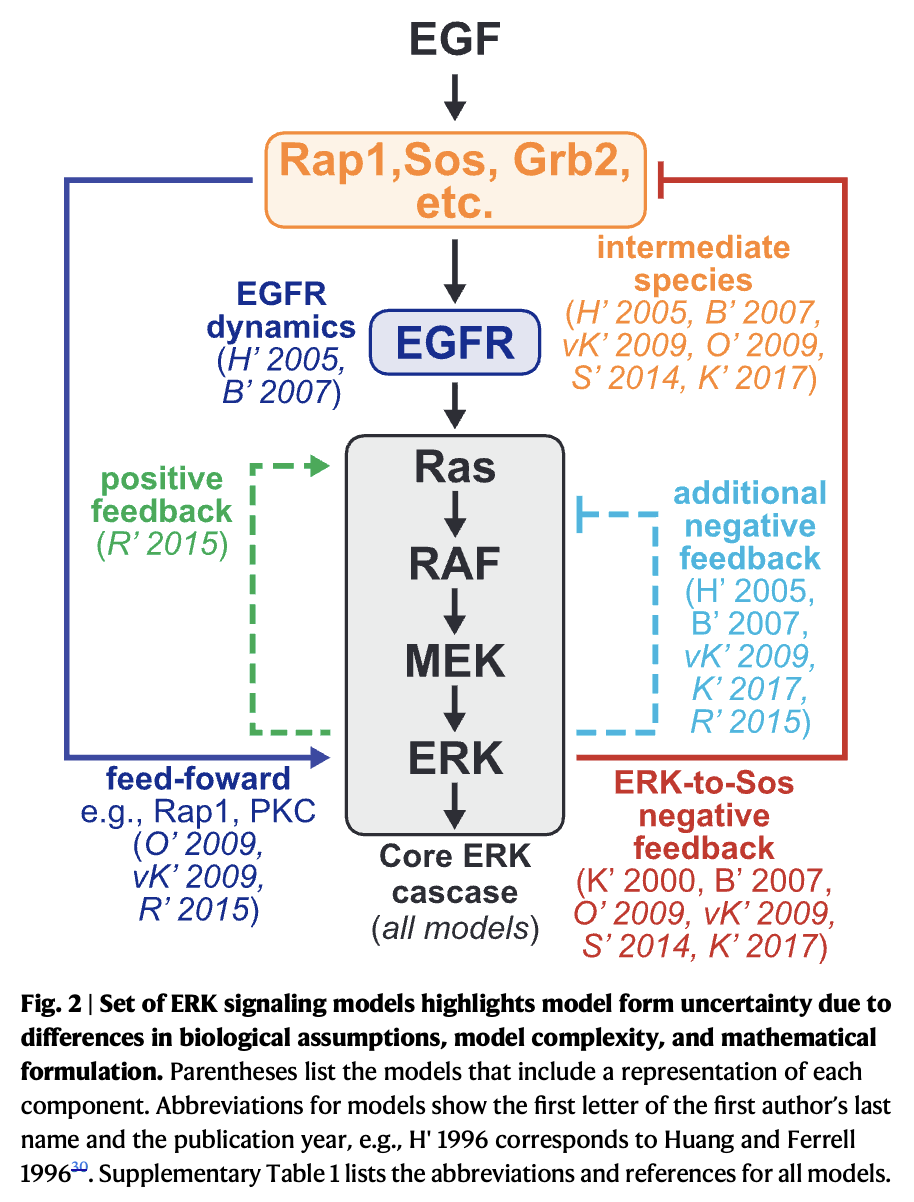
ERK 信号通路在调控多种细胞过程(包括增殖、生长、代谢和分化)中起着关键作用34,35。由于其广泛的重要性,ERK 是被建模最广泛的细胞内信号通路之一36。为了测试多模型推理(MMI)方法,我们选定了一组包含十个核心 ERK 信号网络模型的集合30,37–45。在整个研究中,我们以第一作者姓氏的首字母和发表年份来指代每个模型,例如 Huang 和 Ferrell 于 1996 年提出的模型记为 H’1996。尽管每个模型最初用于回答不同的研究问题,但我们选择这十个模型,是因为它们都完整地描述了从输入到 ERK 激活的 ERK 信号通路。图 2 展示了各个模型所覆盖的表皮生长因子(EGF)至 ERK 的信号网络,并标示出哪些模型包含了特定的结构组分。我们注意到,除了每个模型仅包含完整 ERK 信号网络的子集外,这些模型在机制复杂性和描述反应动力学所用的数学表达形式上也存在差异。例如,H’1996 仅使用质量作用动力学,而 K’2017 则结合了米氏方程(Michaelis-Menten)和希尔方程(Hill-equations)。此外,所有模型都采用单一方程表示 EGF-EGFR 相互作用,只有 H’2005 和 B’2007 采用了详细的多步配体结合和受体内化机制。补充表 1 显示,模型参数数量和状态变量数量的巨大差异反映了模型构建形式上的不同。此外,每个模型的方程均可从原始或二次文献中获得。因此,这些模型在真实世界中体现了建模的不确定性,同时在对 ERK 信号通路的描述上仍具有可比性。基于此,我们认为这组 ERK 模型是测试多模型推理(MMI)的理想案例。
我们先前已表明,先验的结构可识别性分析和全局敏感性分析对于细胞内信号模型的贝叶斯参数估计至关重要12。因此,我们对每个模型进行了局部结构可识别性分析,以确定在参数空间中哪些参数可以被唯一局部识别。接着,我们使用 Morris 筛选法46 对每个模型进行全局敏感性分析,以确定哪些可识别的参数显著影响 ERK 活性的预测结果(见补充图 1)。根据这两项分析的结果,我们将所有不可识别且不具影响的参数固定为文献中的标称值,从而减少了自由参数的数量。补充表 1 列出了经过这些先验分析后剩余的参数数量。我们仅对剩下的自由参数进行估计。
贝叶斯多模型推理提高了 ERK 活性预测的确定性
作为首次测试,我们应用多模型推理(MMI)方法,基于 Keyes 等人27 测得的细胞质 ERK 激酶活性实验数据,预测 EGF 诱导的 ERK 活性。在此,我们旨在更深入理解使用 MMI 进行模型平均如何影响对 EGF 诱导的时变 ERK 活性的预测。Keyes 等人使用一种改进的 ERK 激酶活性报告系统 EKAR4,在亚细胞位置(包括细胞质(图 3a)和细胞膜(补充图 3a))测量了 ERK 活性。我们利用细胞质中的 ERK 活性数据,分别对这十个 ERK 信号模型的参数进行了估计。图 3b 展示了其中四个模型的 ERK 活性预测后验密度(补充图 2a 展示了全部十个模型的密度)。所有十个模型均能较好地定性拟合细胞质 ERK 活性的实验数据。我们认为预测结果中的微小差异源于各模型在形式表达上的不同。
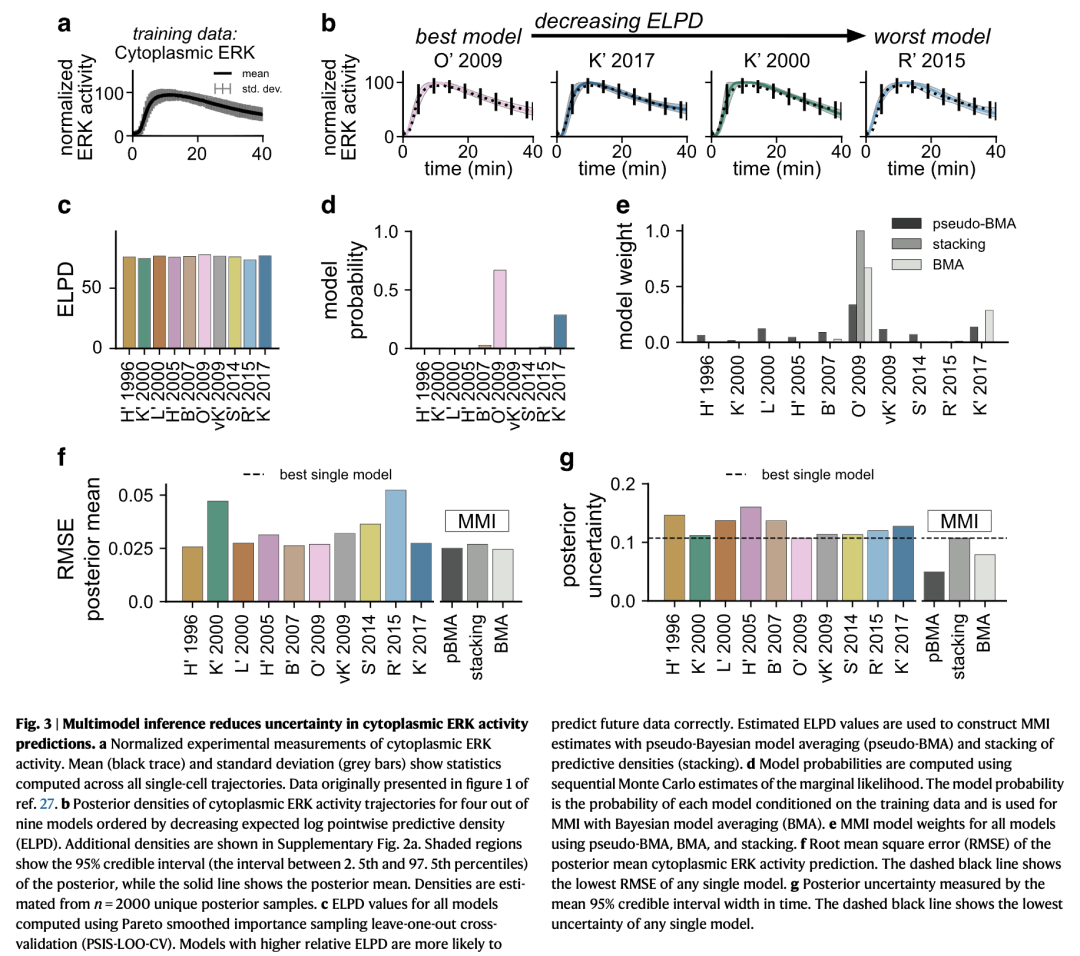
三种 MMI 方法在模型间的权重分配各不相同,但最终的多模型推理预测结果却较为相似。尽管整体趋势相似,但使用 ELPD 值(图 3c)和模型概率(图 3d)对模型进行排序时,两种方法的结果存在差异。我们观察到,对细胞质 ERK 活性拟合效果较好的模型,其 ELPD 值和模型概率均高于拟合较差的模型。伪贝叶斯模型平均(pseudo-BMA)和贝叶斯模型平均(BMA)都为多个模型分配了非零权重,但它们所侧重的模型子集有所不同(图 3e)。
与此同时,堆叠法(stacking)将全部权重都分配给了 O’2009 模型。尽管三种多模型推理(MMI)方法的权重分布不同,但每种方法预测的细胞质 ERK 活性轨迹在定性上均与实验数据相似(补充图 2b)。有趣的是,ELPD 值较高的模型并不一定是结构最复杂或自由参数最多的模型。表现最好的模型 O’2009 和 K’2017 具有中等数量的状态变量,并包含了中间产物和负反馈等组分(见上文)。这表明,要准确捕捉时变的细胞质 ERK 活性,所需的机制复杂性是有限的。
从定量角度来看,不同方法的预测结果存在差异:只有伪贝叶斯模型平均(pseudo-BMA)和贝叶斯模型平均(BMA)提高了预测的确定性。pseudo-BMA 和 BMA 方法的后验均值的均方根误差(RMSE),以及通过后验95%可信区间平均宽度衡量的预测不确定性,均略低于单个最佳模型(图 3f, g)。然而,堆叠法的这些指标与最佳单模型相同,因为它仅对单一模型赋予权重。这些结果表明,对细胞质 ERK 活性的多模型推理预测不仅保持了最佳模型的预测准确性,而且在采用加权平均时还能降低预测不确定性。
为了检验 MMI 是否能改善对其他 ERK 信号数据的预测,我们使用同一组模型,分别基于细胞膜上 ERK 活性的实验测量数据(补充图 3a)以及合成的 EGF-ERK 剂量-反应曲线数据(补充图 4a)重复了 MMI 分析。对于这两个新数据集,不同模型的预测结果各不相同,因此 ELPD 估计值和模型概率也有所不同(补充图 3b–d 和 4b–d)。相应地,每种 MMI 方法也给出了不同的权重分配(补充图 3e 和 4e)。我们注意到,在剂量-反应示例中,一些模型(如 O’2009)未能捕捉到剂量-反应数据的趋势(补充图 4b)。重要的是,这些模型在 MMI 平均中几乎未被赋予权重,因此不会影响 MMI 的最终预测。对于这两个数据集,MMI 预测的误差与表现最佳的单个模型相当。此外,当通过加权平均整合多个模型时,MMI 预测的不确定性低于单个模型的不确定性(补充图 3f, g 和 4f, g)。因此,我们得出结论:除了细胞质 ERK 活性外,MMI 还能有效处理针对细胞膜 ERK 活性和 EGF-ERK 剂量-反应曲线预测中指定模型集合内的模型不确定性。更重要的是,基于所有三个数据集的结果,MMI 预测的误差低于随机选择模型的情况,并且在使用 pseudo-BMA 和 BMA 时表现出更低的不确定性。因此,MMI 能够在多种类型的数据上,提升这组模型预测的确定性。
MMI 提高了对模型形式不确定性的鲁棒性
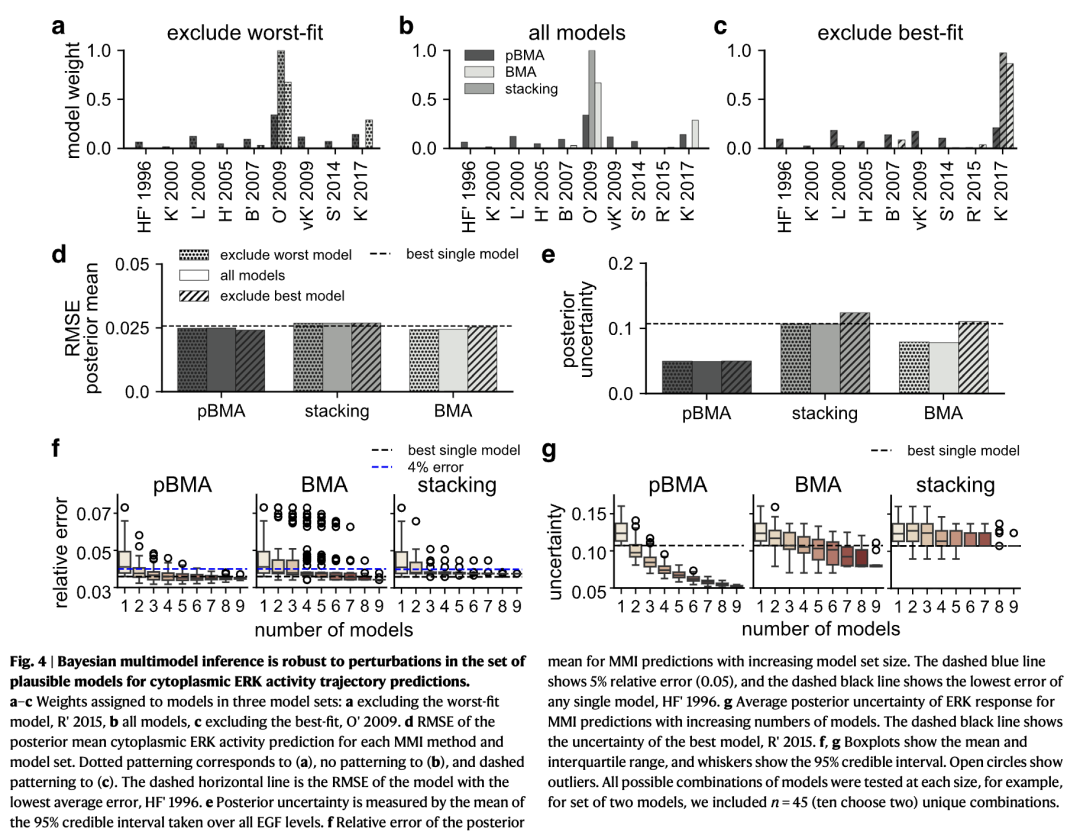
为了理解模型不确定性增加如何影响经模型平均后的多模型推理(MMI)预测,我们系统地改变了模型集合的组成。具体而言,我们探讨了以下三种情况对 MMI 预测性能的影响:(i)添加一个拟合效果差的模型,(ii)移除表现最佳的模型,以及(iii)改变模型集合的大小。
首先,我们发现对模型集合进行单个模型的扰动,通常对 MMI 预测性能的影响较小。为检验其对细胞质 ERK 活性预测的影响,我们首先排除了拟合最差的模型 R’2015(图 3c 中 ELPD 最低),并重新计算了 MMI 权重(图 4a)。将这个拟合差的模型重新加入集合后,MMI 权重几乎没有变化(图 4b),因此对预测误差(图 4d)和后验不确定性(图 4e)的影响极小,因为该不良模型几乎未被赋予任何权重。接着,我们移除了拟合最好的模型 O’2009(ELPD 最高),并重新计算了 MMI 权重(图 4c)。在没有最佳模型的情况下,堆叠法(stacking)将全部权重重新分配给了次优模型 K’2017,而贝叶斯模型平均(BMA)则将权重重新分配给了 K’2017 和 B’2007。伪贝叶斯模型平均(pseudo-BMA)则将原本赋予 O’2009 的权重分散到其他多个模型上。相应地,由于所有模型对细胞质 ERK 活性的预测效果都较好,预测误差仅略有增加。尽管误差略有上升,但移除 ELPD 最高的模型对 MMI 预测的不确定性影响甚微(图 4e)。
我们使用细胞膜数据(补充图 5a–e)和合成的 EGF-ERK 剂量-反应数据(补充图 6a–e)对模型集合进行了相同的单模型扰动,并重复了 MMI 分析。在另外两个数据集上也观察到了类似的结果;然而,在剂量-反应示例中,当我们移除最佳模型时,预测误差增加得更明显,因为该示例中最佳模型与次优模型之间的性能差距更大。因此,这些结果表明,ERK 活性的 MMI 预测对该模型集合的变动具有鲁棒性:添加一个拟合差的模型影响很小,而移除最佳模型也仅导致不确定性和准确性的轻微变化。
接下来,我们研究了实现有效 MMI 所需的模型数量,以及模型集合中模型数量如何影响 MMI 的预测性能。我们针对三个数据集——细胞质 ERK 活性(图 4f)、细胞膜 ERK 活性(补充图 5f)和 EGF-ERK 剂量-反应数据(补充图 6f)——构建了所有可能的模型组合,从包含两个到八个模型的子集(共九个 ERK 模型中选取)。首先,我们发现,随着模型数量的增加,三种数据集上 MMI 预测的平均误差均呈下降趋势,并逐渐接近任意单个模型所能达到的最低误差。特别是,pseudo-BMA 和堆叠法的预测误差下降速度较快,而 BMA 的预测误差即使在模型集合较大时仍保持较高水平。尤其对于 EGF-ERK 剂量-反应曲线的 BMA 预测,相对误差始终集中在 5% 左右,远高于最佳单个模型的误差(补充图 6f)。基于这些发现,我们得出结论:即使仅有两个指定模型,MMI 也能提高正确预测的概率。然而,与 BMA 相比,pseudo-BMA 和堆叠法带来的性能提升更为显著。
模型数量如何影响 MMI 的预测不确定性?对于所有三个数据集,pseudo-BMA 和 BMA 的预测不确定性随着模型集合的增大而降低,但堆叠法并未表现出这种趋势(细胞质 ERK 活性见图 4g;细胞膜 ERK 活性见补充图 5g;EGF-ERK 剂量-反应见补充图 6g)。对于 pseudo-BMA 和 BMA,随着模型数量增加,不确定性逐渐趋近于低于最佳单个模型的水平(即图 4g 中黑色虚线所示)。然而,对于堆叠法,即使使用了三个以上的模型,其不确定性仍与 ELPD 最高的单个模型的预测不确定性相近。因此,我们得出结论:即使模型数量适中,MMI 也能降低不确定性;在本研究的三个示例中,模型数量越多,pseudo-BMA 和 BMA 的不确定性降低越显著。此外,这些结果还表明,即使模型数量增加带来了更大的模型不确定性,MMI 仍能可靠地提高预测的确定性,并生成与最佳单个模型同样准确的预测。
MMI 对训练数据不确定性的增加具有鲁棒性
我们此前已观察到,数据不确定性会显著影响细胞内信号模型的预测不确定性12。此外,训练数据的数量和质量也会影响不同多模型推理(MMI)方法对模型的加权方式22。为了探究 MMI 在存在数据不确定性时的表现,我们通过减少提供给 MMI 的训练数据的数量和质量,模拟了数据不确定性逐步增加的情况。
首先,我们发现,对未来的细胞质 ERK 活性进行预测时,MMI 预测的不确定性低于单个模型的预测。我们通过将原始 40 分钟长的记录分别截断至 10、20 和 30 分钟的时间点(图 5a),改变训练数据的长度,并使用 MMI 预测剩余至 40 分钟的轨迹。我们通过评估轨迹最后 10 分钟(未用于参数估计)的测试误差和不确定性来评价预测性能。与所有单个模型类似,表现最佳的单模型 O’2009 的后验不确定性随着训练数据长度的减少而增加(图 5b 顶部行;其他单个模型的预测见补充图 7)。然而,pseudo-BMA 的预测在所有数据长度下均表现出显著更低的预测不确定性(图 5b 底部行)。从定量上看,MMI 预测的测试误差在所有训练数据长度下均与误差最低的单个模型相当(图 5c),而 MMI 预测的测试不确定性则显著低于最佳单个模型(图 5d)。需要注意的是,在 10 分钟和 20 分钟的数据长度下,L’2000、K’2000 和 H’2005 的预测不确定性低于 MMI 预测,但它们的预测误差极高,几乎所有模拟都预测在 40 分钟时达到最大激活(补充图 7)。我们还使用细胞膜数据重复了相同的模拟,观察到了与细胞质数据相似的趋势(补充图 8a–e)。综上,这些结果表明,与仅使用单个模型相比,MMI 能够在使用更短训练数据的情况下,提高对未来 ERK 活性预测的确定性和准确性。
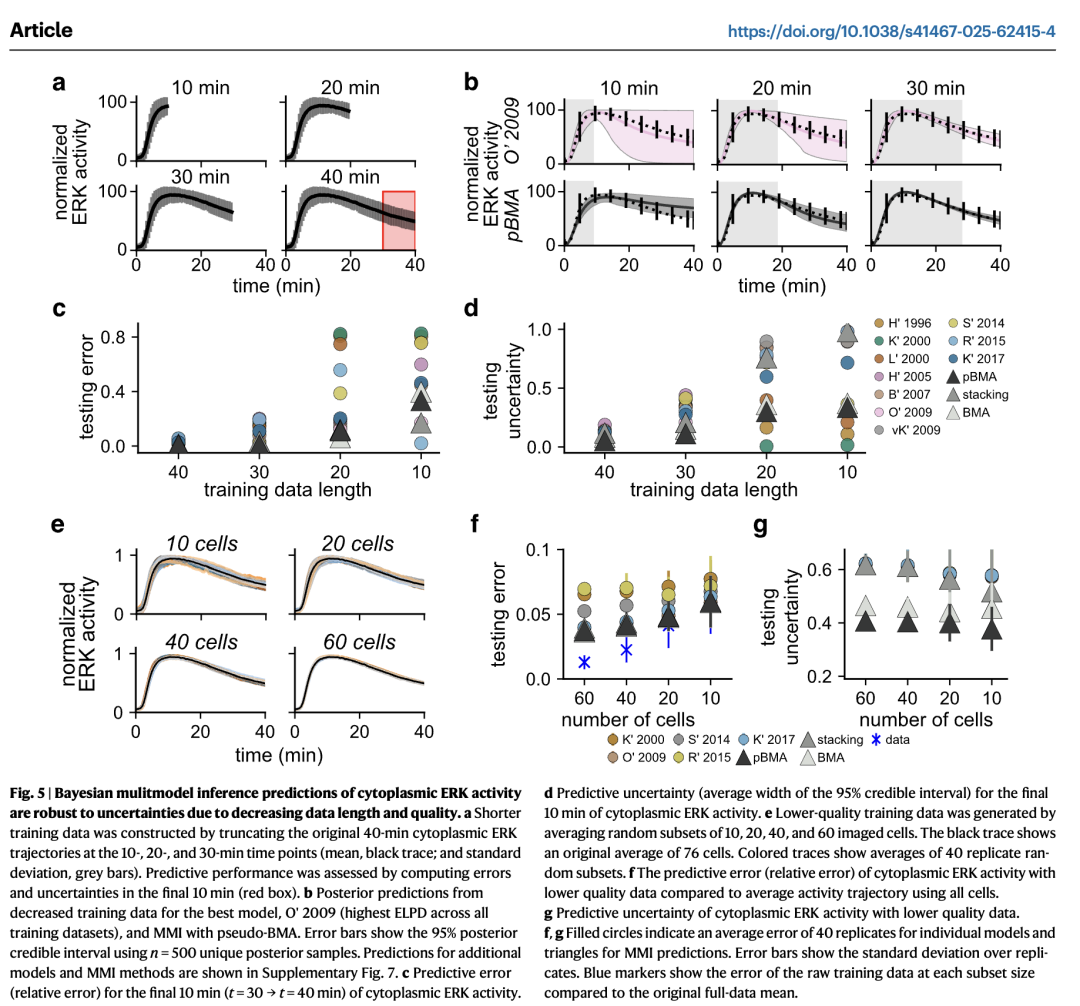
其次,MMI 预测在低质量数据下的预测误差和不确定性也低于单个模型的预测。我们通过减少用于生成归一化数据的单细胞记录数量,来生成质量较低的训练数据。具体而言,我们从 76 条原始的细胞质 ERK 活性单细胞记录中,随机抽取了 40 组包含 10、20、40 和 60 个细胞的子集,并计算了细胞层面的平均值和标准差(图 5f)。使用较少细胞数(10 或 20 个)生成的数据质量低于使用较多细胞数(40 或 60 个)的数据,因为细胞数较少的数据与原始 76 细胞数据集相比误差更大,且不确定性波动更明显(补充图 9)。为减轻重复参数估计带来的计算负担,我们从十个模型中选取了五个推理时间最短的模型(K’2000、O’2009、S’2014、R’2015 和 K’2017)来构建 MMI 估计(补充表 3)。预测的测试误差以模型预测相对于原始 76 细胞数据集的相对误差计算,结果显示,MMI 预测比大多数单个模型更准确(图 5g)。此外,BMA 和 pseudo-BMA 的 MMI 预测在所有子集大小下均表现出更低的预测不确定性(图 5h)。我们同样使用细胞膜数据重复了这些模拟,结果同样表明 MMI 对低质量数据具有鲁棒性(补充图 8f, g)。基于这些结果,我们得出结论:与单个模型预测相比,MMI 预测对因数据质量下降引起的数据不确定性更具鲁棒性。
亚细胞ERK活性依赖于Rap1和负反馈
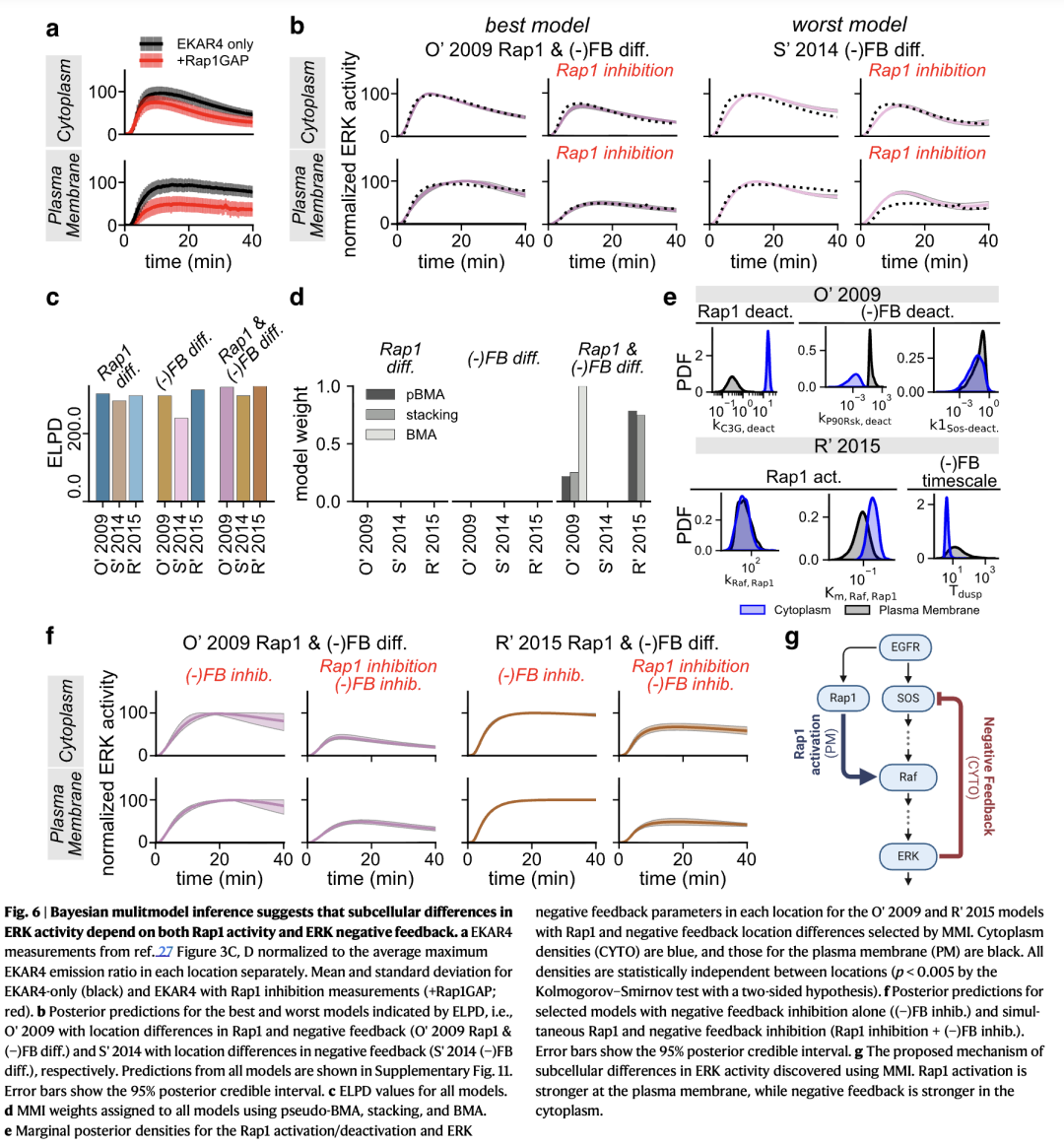
Keyes等人先前揭示了细胞质与细胞膜上ERK信号在时空上的差异27。具体而言,作者观察到,与细胞质相比,细胞膜上的ERK活性持续时间更长(图6a,黑色轨迹;数据源自参考文献27的图1C和1D)。此外,Keyes等人还表明,细胞膜上的ERK活性强烈依赖于非经典ERK激活因子Rap1,而细胞质中的活性则不依赖(参考文献27的图3C和3D)。在此,我们使用多模型推理(MMI)来探究模型参数的亚细胞特异性差异是否能够预测ERK活性的亚细胞变异,并筛选出能准确解释实验数据的模型。
最初,我们假设模型参数的亚细胞特异性概率密度可以解释Keyes等人27观察到的亚细胞特异性ERK活性。在前几节中,我们发现当所有参数在两个区室间可独立变化时,相同的模型能够较好地预测细胞质和细胞膜上的ERK活性(见上文)。补充图10突出了每个模型中最显著的亚细胞特异性参数估计值。例如,在Raf失活、EGFR动力学、ERK激活以及总激酶浓度方面均存在亚细胞差异。然而,由于我们允许所有模型参数在区室间独立变化,这些发现仅说明哪些亚细胞参数差异可能导致对ERK活性更准确的预测,但并未揭示哪些特定的独立参数组合是捕捉实验数据所必需的。
为了探究哪些参数组合能够解释亚细胞特异性ERK活性,我们使用贝叶斯多模型推理(MMI)评估了所有可能的区室间参数变化组合。具体而言,我们聚焦于O’2009、S’2014和R’2015这三个模型,因为它们全面涵盖了核心ERK网络。此外,将分析限制在三个模型内可确保计算的可行性。在这三个模型中,我们观察到控制Raf失活、ERK磷酸化、ERK去磷酸化、ERK负反馈以及Rap1活性的参数在不同区室间存在显著差异(补充图10及补充材料)。因此,我们允许相关参数的组合在区室间变化。由于细胞膜上ERK活性对Rap1的依赖性是Keyes等人27的关键发现,我们在S’2014和R’2015模型中引入了一个类似Rap1的激活因子,使所有三个模型都能用于预测Rap1抑制后的ERK活性。为此,我们假设活化的EGFR激活Rap1,进而激活下游的Raf激酶,并且Rap1的激活不受活化ERK的负反馈调节47。随后,我们进行了94组参数估计,允许相关参数的每种组合在区室间独立变化或保持全局一致。我们将每个假设下的模型拟合到仅使用EKAR4以及EKAR4结合Rap1抑制剂(Rap1GAP)的数据(图6a)。为模拟Rap1抑制,我们将总Rap1浓度设为零。
接下来,我们使用MMI比较了哪些参数组合最能捕捉亚细胞ERK活性数据。MMI对各种参数组合赋予了不同权重(补充表4列出了权重最高的前十种和最低的十种组合)。有八种参数组合的权重大于0.01。这些组合均允许ERK磷酸化和/或去磷酸化参数在区室间变化。其他包含的亚细胞特异性参数组合还包括Rap1激活、ERK负反馈以及Raf失活(补充表4)。识别出的控制ERK磷酸化的亚细胞特异性参数可能表明模型中未包含的物种负责亚细胞特异性动力学,例如潜在的串扰或其他反馈机制。然而,也有可能这些参数只是最容易估计的。因此,这些亚细胞差异可能并不代表生物学上真实的机制,因为系统生物学模型已知能够拟合广泛的数据48。
为进一步明确能够解释实验数据的亚细胞特异性参数,我们使用MMI比较了一组更小的、基于已知生物学真实性的假设,以确定哪些参数影响亚细胞特异性ERK活性27,47,49。具体而言,我们重点关注非经典ERK激活因子Rap1和ERK负反馈。我们假设Rap1信号在不同区室间存在差异,因为Keyes等人27发现细胞膜上的ERK活性依赖于Rap1,而细胞质中的活性则不依赖(参考文献27的图3)。此外,我们假设ERK负反馈在不同区室间可能存在差异,因为先前的研究表明,ERK负反馈的强度可以驱动持续性与瞬时性ERK活性的差异47,49。为检验这些关于亚细胞特异性信号的假设,我们进行了九组参数估计,其中仅允许Rap1参数(Rap1 diff.)、仅允许ERK负反馈参数((−)FB diff.),或同时允许Rap1和ERK负反馈参数(Rap1 & (−)FB diff.)在不同位置独立变化。与Rap1或ERK负反馈无关的参数被假定在区室间不变,并对这些参数估计单一的概率密度。
Rap1信号和ERK负反馈的亚细胞特异性差异对于预测ERK活性的亚细胞变异性都是必需的。从定性上看,仅允许ERK负反馈参数在区室间变化的模型((−)FB diff.)对数据的预测较差,而允许Rap1参数变化的模型(Rap1 diff.)对数据的预测更准确。同时允许Rap1和ERK负反馈参数变化的模型(Rap1 & (−)FB diff.)似乎对数据的预测效果最好(图6c和补充图11)。预测的ELPD值显示,Rap1 & (−)FB diff.模型确实对数据的预测效果最好,其次是Rap1 diff.和(−)FB diff.模型(图6c)。此外,所有三种MMI方法仅对同时允许Rap1和负反馈差异的假设赋予权重(图6d)。有趣的是,pseudo-BMA和堆叠法同时选择了基于O’2009和R’2015的模型,而BMA仅选择了基于O’2009的模型。这表明,除了细胞膜上的Rap1依赖性外,亚细胞特异性的ERK负反馈也在亚细胞特异性ERK活性中发挥作用。
有趣的是,在MMI选出的假设中,Rap1的激活在细胞膜上更强,而ERK负反馈在细胞质中更强,这由相关参数的边缘后验密度表明(图6e)。具体而言,对于O’2009模型,Rap1激活因子C3G的失活速率(k1C3G-deact.)在细胞膜上较低;对于R’2015模型,Rap1–Raf平衡常数激活速率(Km,Raf,Rap1)在细胞膜上较低。此外,参数估计表明,细胞质中的ERK负反馈强于细胞膜。这些结果使我们得出结论:更强的Rap1活性驱动了细胞膜上持续的ERK活性,而更强的负反馈是细胞质中瞬时ERK活性所必需的。事实上,抑制ERK负反馈几乎消除了ERK活性的亚细胞差异(图6f)。在细胞质中抑制ERK负反馈导致两个模型中的ERK激活,从而丧失了亚细胞特异性信号。然而,在细胞膜上,负反馈抑制对ERK活性影响甚微。因此,这些结果表明,ERK负反馈和Rap1活性对于亚细胞特异性ERK活性是必需的。
在此,MMI使我们能够从一组预选的、生物学相关的假设中选择最符合数据的假设。这些发现表明,细胞质ERK活性依赖于ERK负反馈,而细胞膜ERK活性依赖于Rap1。基于MMI选出的假设,我们提出了一个新的ERK负反馈和Rap1依赖性模型,用于解释ERK活性的亚细胞变异性(图6f)。
讨论
在本研究中,我们展示了贝叶斯多模型推理(MMI)在ERK信号通路中的一项新颖应用。MMI使我们能够应对系统生物学中的模型形式不确定性,特别是在存在同一系统的多个模型时。我们比较了三种MMI方法——贝叶斯模型平均(BMA)、伪贝叶斯模型平均(pseudo-BMA)和预测密度堆叠法(stacking)。基于十个ERK信号模型,我们将贝叶斯多模型推理应用于两个基本任务:模型平均和模型选择。我们发现,用于模型平均的MMI通过整合多个模型降低了预测不确定性(见“结果”部分“贝叶斯多模型推理提高了ERK活性预测的确定性”),保持了最佳单个模型的预测准确性,并且对模型集合的扰动具有鲁棒性(见“结果”部分“MMI提高了对模型形式不确定性的鲁棒性”)。我们还发现,与单个模型相比,MMI预测对额外的数据不确定性更具鲁棒性,并能实现更优的样本外预测(见“结果”部分“MMI对训练数据不确定性的增加具有鲁棒性”)。最后,在用于模型选择时,MMI使我们能够系统地比较大量假设,并选择与数据最一致的假设(见“结果”部分“亚细胞ERK活性依赖于Rap1和负反馈”)。尽管我们的研究基于以ERK信号通路为例的一系列分析,但本工作的启示——即利用多个模型可以提升预测性能并增强鲁棒性——很可能对整个系统生物学领域及更广泛的建模社区产生广泛影响。
通常,人们会根据模型的生物学假设和适用范围来选择一个“好”模型,而不评估其预测性能。相比之下,我们发现贝叶斯多模型推理在预测方面具有多个优势。
多模型推理(MMI)通过基于预测性能对模型进行加权,提高了做出准确预测的可能性。此外,我们发现,当对多个模型的预测结果进行平均时,MMI的预测不确定性会降低。因此,由参数估计和模型结构引起的整体不确定性贡献将减少。后验预测模拟(见“方法”部分公式(8))反映了总不确定性,其中包括后验不确定性和数据不确定性13。需要指出的是,本文仅展示了后验密度,以便更清晰地突出不同模型之间的差异。这一点在模型用于预测未来ERK活性时尤为明显(见“结果”部分“MMI对训练数据不确定性的增加具有鲁棒性”)。最后,我们发现MMI对模型设定错误和错误假设具有鲁棒性,因为它会忽略来自表现最差模型的预测。例如,如果我们仅选择使用O’2009模型(这是一个合理且有充分依据的选择)来预测EGF-ERK剂量-反应关系,预测结果将非常糟糕(见补充图4);然而,MMI几乎未给该模型赋予权重。这些发现表明,MMI通过利用所有用户指定的模型来考虑模型不确定性,从而提高预测的确定性。
是否存在适用于系统生物学的最佳贝叶斯多模型推理方法?在本研究中,我们发现伪贝叶斯模型平均(pseudo-BMA)——只要使用任何贝叶斯推理方法即可广泛应用18——产生了最准确且最确定的预测。堆叠法(stacking)倾向于仅选择ELPD最高的模型,在我们的示例中尤其如此,除非像“结果”部分“亚细胞ERK活性依赖于Rap1和负反馈”中那样包含多种数据。需要强调的是,当所有模型的预测结果相似时,堆叠法可能仅对单一模型赋予权重50。最后,我们发现,尽管贝叶斯模型平均(BMA)在模型平均方面有效,但在某些情况下可能显著降低预测不确定性的同时,反而使预测误差超过最佳单个模型(例如,见补充图3)。此外,BMA需要使用特定的采样器(如序列蒙特卡洛SMC)或额外计算来估计边缘似然19,22。尽管pseudo-BMA在模型平均方面表现良好,且比BMA更具普适性,但两种方法相比单一模型均展现出优势。此外,在模型选择方面,我们发现结合多种MMI方法进行模型选择应用,可增强对所选模型或假设的信心(例如,“结果”部分“亚细胞ERK活性依赖于Rap1和负反馈”)。因此,基于我们的研究结果,我们认为pseudo-BMA是贝叶斯多模型推理的一种良好通用方法,并建议在模型选择时使用多种方法。我们还注意到,直接比较贝叶斯与频率学派方法有助于加深对现有MMI工具集的理解。
值得注意的是,本研究分析的三种MMI方法之外还存在其他替代方法。首先,我们选择聚焦于贝叶斯方法,而排除了使用赤池信息准则(AIC)或贝叶斯信息准则(BIC)等指标的频率学派方法14,18,因为贝叶斯方法提供的不确定性量化更适合系统生物学中数据有限的情况。在贝叶斯文献中,随机搜索变量选择51、概率密度融合24和可逆跳转MCMC52等方法也被提出用于多模型推理(更多方法参见文献19和18中的引用)。诸如集成估计(ensemble estimation)53等方法可产生与MMI类似的预测。因果推断54以及促进稀疏性的推断方法55–58可更直接地从数据中选择模型或机制。将基于MMI的模型选择与替代的数据驱动方法进行比较,有望进一步扩展系统生物学可用的工具。
进行有效的多模型推理需要多少数据?尽管我们发现MMI估计对训练数据量具有鲁棒性,但不同应用场景和模型集合所需的最小数据量可能各不相同。在本研究中,我们使用了直接测量感兴趣量(QoI)的数据;然而,若使用不直接测量QoI的数据进行MMI,可能会更具挑战性,或导致预测不确定性更高。总体而言,我们注意到MMI预测的确定性和准确性直接依赖于单个模型预测的质量,而后者已知会随着数据量的增加而提升(例如,参见文献12)。评估可用数据量如何影响MMI预测的一种可能方法是纳入模型权重的不确定性。例如,Yao等人22利用贝叶斯自助法(Bayesian bootstrap)通过抽样可能的pseudo-BMA模型权重的近似分布来评估模型权重的不确定性。此外,本研究中的MMI基于单一数据集评估预测性能的指标,而整合多个数据集可能进一步提升MMI预测的置信度和准确性。此类工作可能需要多数据多模型推理方法,可借鉴元建模(metamodeling)的思想来对数据集和模型进行加权。
未来MMI在系统生物学中的发展应聚焦于采用计算高效的参数估计方法,并探索学习时变MMI权重的技术。MMI需要对每个指定模型重复进行贝叶斯参数估计,这可能给建模研究带来巨大的计算负担(例如,补充表3中的推理运行时间)。加速贝叶斯估计的方法,如变分推断(variational inference)59或拉普拉斯近似(Laplace approximation)28,可在提供近似不确定性量化的同时,使MMI适用于参数估计成本高昂的模型。此外,本文所呈现的MMI框架中,各模型的权重不随时间变化。然而,在存在显著时变特征的场景中(如细胞命运转变或发育过程),驱动生物过程的潜在机制可能发生改变,因此最适用的模型也可能随时间变化。在这种情况下,对MMI框架的扩展方法,如动态BMA16或多模型序列数据同化60,可能学习到随时间变化的模型权重,从而使MMI估计器能够随时间调整。
随着数学模型在研究生物系统中持续发挥关键作用,严谨地考虑模型假设变得至关重要。尽管建模的黄金标准之一是开发能完美捕捉生物学现实的模型,但我们对生物过程理解的局限性迫使我们依赖简化假设,以构建更易处理的简化模型。此外,开发和使用复杂、高度逼真的模型需要具备收集大量高质量数据的能力,以支持参数估计并做出准确预测。鉴于我们常常面临知识不完整和数据有限的情况,我们必须能够做出兼顾并适应建模假设相关不确定性的预测。因此,在这些情况下,我们提出,贝叶斯多模型推理提供了一种严谨的方法来处理模型不确定性,有望在存在同一系统的多个模型时,提升系统生物学的预测能力,并推动未来的生物学发现。此外,当我们拥有大数据集时,MMI可作为开发详细、符合生物学实际模型的起点,通过确定与数据最一致的简单模型组分来指导建模。总之,我们认为MMI是一种在系统生物学中应对建模假设的强大方法,能够提高预测的确定性,并为未来的模型开发提供指导。
方法
标准化的EGF输入
为了在不同模型间统一EGF输入,我们修改了所有模型的表达形式,将EGF作为一个状态变量纳入其中。然后,我们将EGF的时间导数设为零,以模拟持续的EGF刺激,这一假设意味着模型中的反应不会消耗细胞外的EGF池。此外,我们将所有EGF刺激的浓度统一定义为纳摩尔(nM)。为将ng/mL或pg/mL单位的浓度转换为nM,我们假设EGF的分子量为6,048 g/mol⁶¹。此外,为将nM转换为分子数/细胞,我们假设细胞体积为1纳升(nL)⁶²。最后,在包含其他生长因子的模型中,我们将相应受体的浓度设为零。具体而言,在Birtwistle等人2007⁴⁰的模型中,我们固定了H、E3和E4的浓度;在von Kreigsheim等人2009⁴²的模型中,我们固定了NGF和NGFR的浓度。
实验数据预处理
Keyes等人通过活细胞荧光显微镜实验报告了所有EKAR测量值,即YFP/CFP发射比值R(t)²⁷。在“结果”部分“贝叶斯多模型推理提高ERK活性预测的确定性”中,我们对每条单细胞轨迹进行了归一化处理:减去该细胞轨迹的最小值,再除以该轨迹最大值与最小值之差。此外,在“结果”部分“亚细胞ERK活性依赖于Rap1和负反馈”中,我们将归一化的EKAR4数据定义为 ~R(t) := R(t)/Rmax,其中R(t)为EKAR4的发射比值,Rmax为所有细胞(包括有无Rap1抑制)在时间上最大发射比值的均值。这种归一化方式保留了ERK信号的亚细胞位置特异性差异以及Rap1抑制的影响。所有原始数据均为单个细胞的测量值;因此,为了得到一个具有不确定性估计的单一数据集,我们计算了所有细胞的平均值和标准差。我们发现归一化在Rap1抑制数据中引入了较大的不确定性,因此在构建似然模型时,我们将数据的标准差设为直接计算值的一半。为确保数据与模型预测在同一尺度上,我们同样将模型预测的活性ERK值按其轨迹的最大值进行归一化。
合成数据生成
我们生成了两个符合实验实际情况的合成数据集用于分析多模型推理(MMI)。首先,使用H’1996模型,我们重现了参考文献30图2B中所示的EGF-ERK剂量-反应曲线:将输入EGF浓度从0.001 nM到0.106 nM分为10个水平,计算相应的稳态ERK活性。EGF输入浓度范围也与参考文献63中的实验相似。具体而言,我们将每个响应值相对于所有输入水平中的最大值进行归一化,并以“最大ERK活性的百分比”表示。此外,我们假设每次测量均受到均值为零、独立的高斯测量误差影响,标准差为0.1,这与实验中观察到的测量不确定性相似³⁰,⁶³。为考虑不同模型中可用ERK总量的差异,并确保模型预测与数据在同一尺度上,我们通过将每条预测的剂量-反应曲线归一化到该曲线中的最大ERK活性,来计算“最大ERK活性的百分比”。
贝叶斯参数估计与序列蒙特卡洛
在本节中,我们简要概述贝叶斯参数估计方法。关于系统生物学背景下的更多细节,请参见文献11,12;关于一般理论的更深入内容,请参见文献10,13。

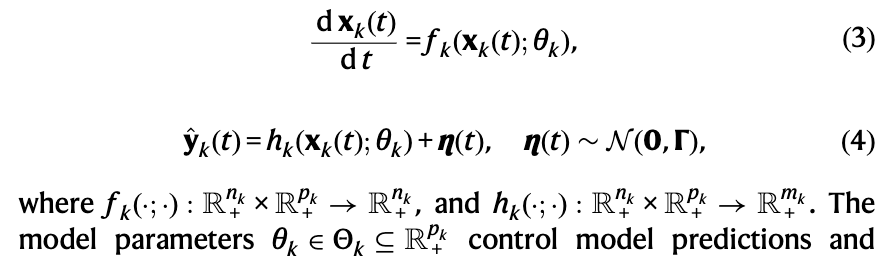


我们使用SMC从后验密度中采样,因为它能够在不进行额外计算的情况下估计边缘似然64,65。SMC是一种基于粒子的采样器,它通过一种退火策略,依次将先验样本逐步调整为后验样本。在每次采样阶段,粒子会根据重要性采样步骤被赋予权重并发生变异。在最终阶段,分配给粒子的权重对应于边缘似然,可以对其进行平均以提供边缘似然的估计值。我们使用PyMC Python包提供的实现,其中采用独立的Metropolis-Hastings转移核。此外,我们将correlation_threshold参数设为0.01,threshold参数设为0.85。除非另有说明,我们运行四个独立的SMC链,每条链至少包含500个后验样本。
通过集成模拟进行前向不确定性传播
给定来自后验密度的 S 个样本,我们进行集成模拟,将不确定性向前传播至模型预测。具体而言,我们使用每个后验样本 S 求解常微分方程(ODE)模型,生成一组 ODE 解,从而得到相应的预测密度。我们使用三种重要的预测密度来评估预测不确定性。首先,感兴趣量(QoI)的后验前推密度(也称为 QoI 的预测密度)定义为:

贝叶斯模型平均



伪贝叶斯模型平均
伪贝叶斯模型平均(Pseudo-Bayesian model averaging)根据模型对未来数据的预期预测性能来对模型进行加权,该性能通过“期望对数逐点预测密度”(ELPD)进行衡量。类似于使用AIC代替ELPD的赤池型加权法14,我们将标量形式的伪贝叶斯模型平均(pseudo-BMA)权重定义为:
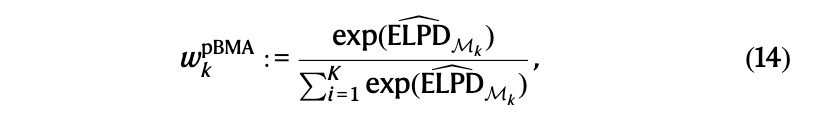

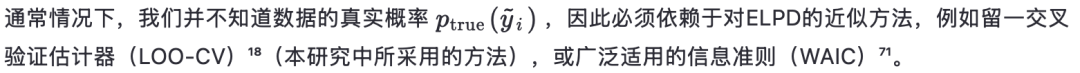
对ELPD最简单的近似称为“样本近似的逐点对数预测密度”(LPD),它仅仅是将公式(17)在所有数据点上求和:


在本研究中,我们使用 ArviZ Python 库⁷³ 来计算 PSIS-LOO-CV 及由此得到的模型权重。给定一个包含各模型对数似然样本的推断数据对象字典,我们使用 Arviz.compare() 函数,将 ic 参数设置为 loo,并将 method参数设置为 BB-pseudo-BMA。所有计算均采用默认设置。为确保 PSIS-LOO-CV 能准确估计 LOO-CV 估计量,我们将 PSIS-LOO-CV 与直接留一交叉验证(LOO-CV)在 S’2014 模型上进行了比较,使用的是合成的剂量-反应数据(补充图 4a)。值得注意的是,PSIS-LOO-CV 和 LOO-CV 的 ELPD 预测值相差在百分之五以内,分别为 -5.69 和 -5.43。

预测密度的堆叠



结构可辨识性分析 我们进行先验局部结构可辨识性分析,以在给定可观测变量的情况下,将未知模型参数减少为具有唯一值的参数。关于结构可辨识性分析的背景和数学细节,我们建议读者参考文献80,81。我们选择进行局部分析而非全局分析,这与我们之前的工作¹²中所建议的不同,因为对于较大的模型(如H' 2005 和 B' 2007),计算全局可辨识性在计算上不可行。具体来说,我们使用了文献81中提出的结构可辨识性分析方法,该方法已通过Julia编程语言中的StructuralIdentifiability.jl软件包实现。我们对assess_local_identifiability()函数使用默认设置,并将所有模型的正确性概率设为p = 0.99,除了Birtwistle等人于2007年提出的模型⁴⁰,对于该模型我们将p设为0.95以提高计算效率。此外,由于软件限制,我们排除了指数项中的任何参数进行可辨识性分析。如果这些参数是整数,我们将其固定为其标称值;否则,我们将其设为1.0。具体而言,对于Hornberg等人于2005年提出的模型³⁹,我们将n固定为1.0;而对于von Kreigshelm等人于2009年提出的模型⁴²,我们将k₅₇、k₆₁、k₆₄、k₆₆、k₇₀和k₇₂固定为1.0。
全局敏感性分析 我们采用Morris筛选法进行全局敏感性分析⁸²,⁸³。首先,我们假设所有参数独立变化,且取值范围为[10⁻² × θ_nominal, 10² × θ_nominal](标称值列于补充材料中)。然后,我们使用稳态激活的ERK浓度作为感兴趣量(QoI),用于预测在标称参数值下持续ERK激活的模型,即H' 1996、O' 2009、S' 2014、R' 2015和K' 2017;或者使用最大激活的ERK浓度作为QoI,用于预测瞬时激活的模型,即K' 2000、L' 2000、H' 2005、B' 2007和vK' 2009。此外,我们假设EGF浓度在所有模型中均固定为0.1 nM。为了执行Morris筛选,我们使用Python中的SALib软件包⁸⁴,⁸⁵,采用Morris采样器方法(SALib.sample.morris.sample())和分析方法(SALib.sample.morris.analyze()),并使用默认设置。对于除H' 2005和B' 2007之外的所有模型,我们在每个参数方向上抽取256个样本;对于H' 2005和B' 2007,我们分别使用30和10个样本。Morris筛选提供两个敏感性度量:元素效应绝对值分布的均值μ* 和元素效应分布的标准差σ*。有关更多细节,请参见文献46,83。我们假设当μᵢ*/max{μᵢ*} > 0.1 或 σᵢ/max{σᵢ*} > 0.1时,参数对ERK激活具有显著影响。
误差与不确定性指标 我们使用均方根误差(RMSE)或相对误差来衡量预测误差。给定一个预测值ŷ和一个参考值(真实值)y,RMSE定义如下:


概率密度的统计比较我们使用Kolmogorov-Smirnov检验(K-S检验)或Mann-Whitney U检验(双侧假设)来比较概率密度。所有统计比较均使用Scipy Python库进行。
原文链接:https://www.nature.com/articles/s41467-025-62415-4
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-09-04,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号