文献分享--血管STING激活促进自然杀伤细胞在小细胞肺癌中的抗肿瘤免疫
原创
文献分享--血管STING激活促进自然杀伤细胞在小细胞肺癌中的抗肿瘤免疫
原创
追风少年i
发布于 2026-03-09 11:03:10
发布于 2026-03-09 11:03:10
作者,Evil Genius
每年的大约1-5月份左右,都是新技术、新思路、高分文章产出的高峰期,而11-12月一般是平静期,不知道这是不是跟国人的习惯有关。
读文献不仅仅是看看其中主要的研究策略,也看看前沿的研究成果都有什么。
今天我们分享文献,visium的运用文章

知识积累
近期研究揭示了神经内分泌肿瘤类型(如小细胞肺癌和神经母细胞瘤)中维持"冷"免疫表型的表观遗传机制。这类肿瘤免疫微环境(TIME)的典型特征包括免疫细胞稀少/排斥、免疫抑制性细胞因子以及肿瘤细胞STING基因沉默。与其他高突变癌种相比,SCLC对免疫检查点阻断(ICB)的响应率也较低。
肿瘤相关血管是免疫效应细胞进入TIME的主要物理屏障。内皮细胞(EC)通过诱导性表达选择素(E-、P-、L-选择素)、细胞间黏附分子1(ICAM-1)和血管细胞黏附分子1(VCAM-1)等配体,调控免疫细胞向组织微环境的外渗过程。虽然EC基础表达此类黏附分子几乎缺失,但炎症刺激可快速上调其表达。通过TIME内在信号或外源性刺激激活此类血管黏附分子以促进原位或过继转移免疫细胞浸润,已成为当前研究热点。
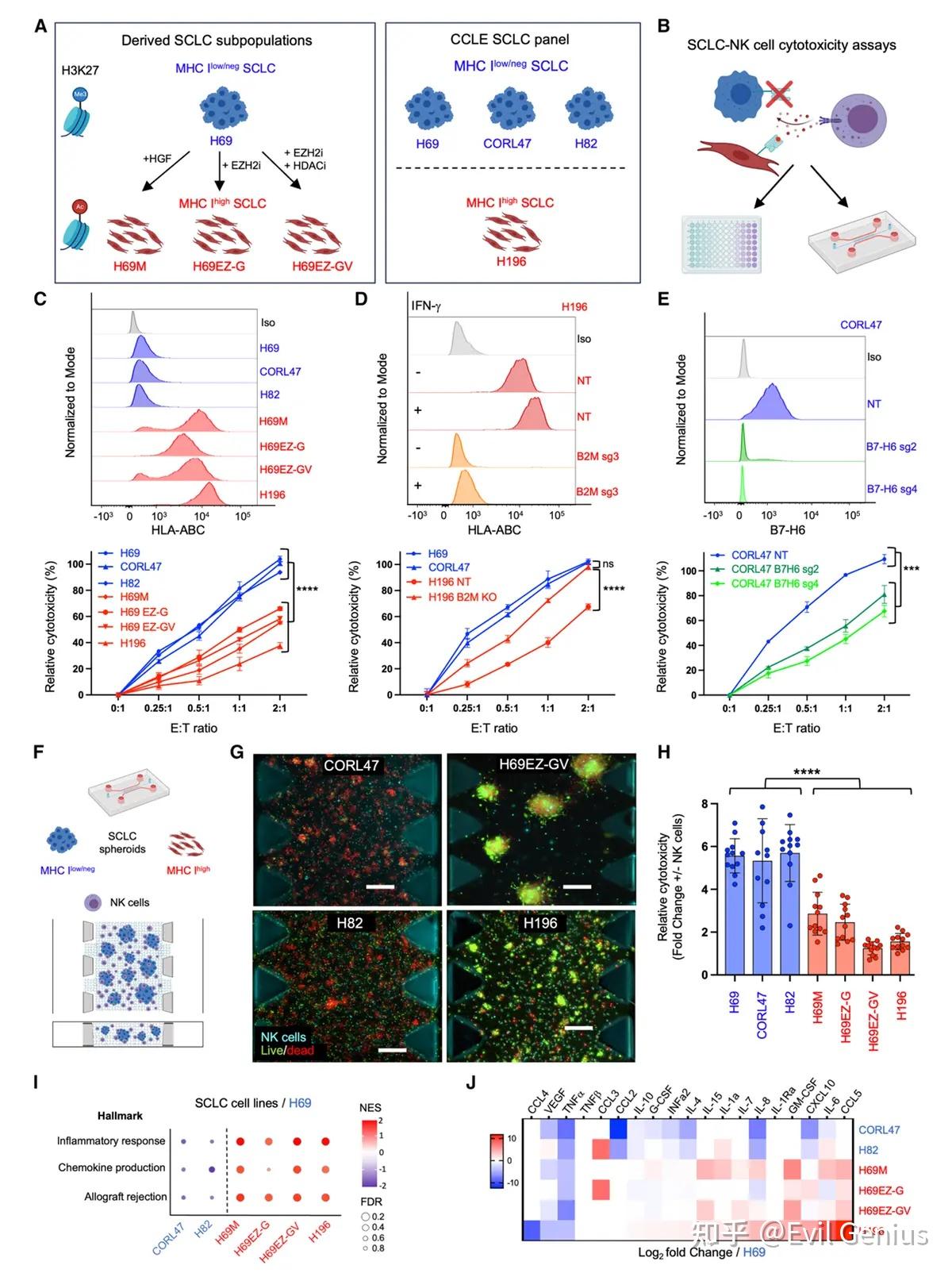
结果1、神经内分泌MHC-I低表达/阴性小细胞肺癌在体外对NK细胞介导的细胞毒性敏感
NE MHC-I低表达/阴性SCLC细胞系对NK细胞介导的细胞毒性高度敏感,而非NE MHC-I高表达SCLC细胞系则相对抵抗。
B7-H6作为SCLC中NK细胞激活性配体的作用。
为在更接近生理相关性的SCLC-NK细胞互作体系中验证上述发现,构建了三维微生理模型,将SCLC球状体与NK细胞在胶原基质中共培养,从而开展3D细胞毒性实验。与二维培养结果一致,NE MHC-I低表达/阴性SCLC细胞在直接混合时对NK细胞杀伤高度敏感,而非NE MHC-I高表达SCLC细胞系则相对抵抗。综上,这些结果表明当空间临近时(包括在更接近三维肿瘤环境的培养模型中),NE MHC-I低表达/阴性SCLC对NK细胞介导的细胞毒性敏感。
尽管直接接触时对NK细胞敏感,我们推测同样缺乏STING的NE MHC-I低表达/阴性SCLC可能无法招募NK细胞,因为已知STING表观遗传沉默会损害IRF3驱动的T/NK细胞趋化因子表达。尽管这些肿瘤对NK细胞毒性敏感,但其可能缺乏效应免疫细胞趋化所需的必要信号。
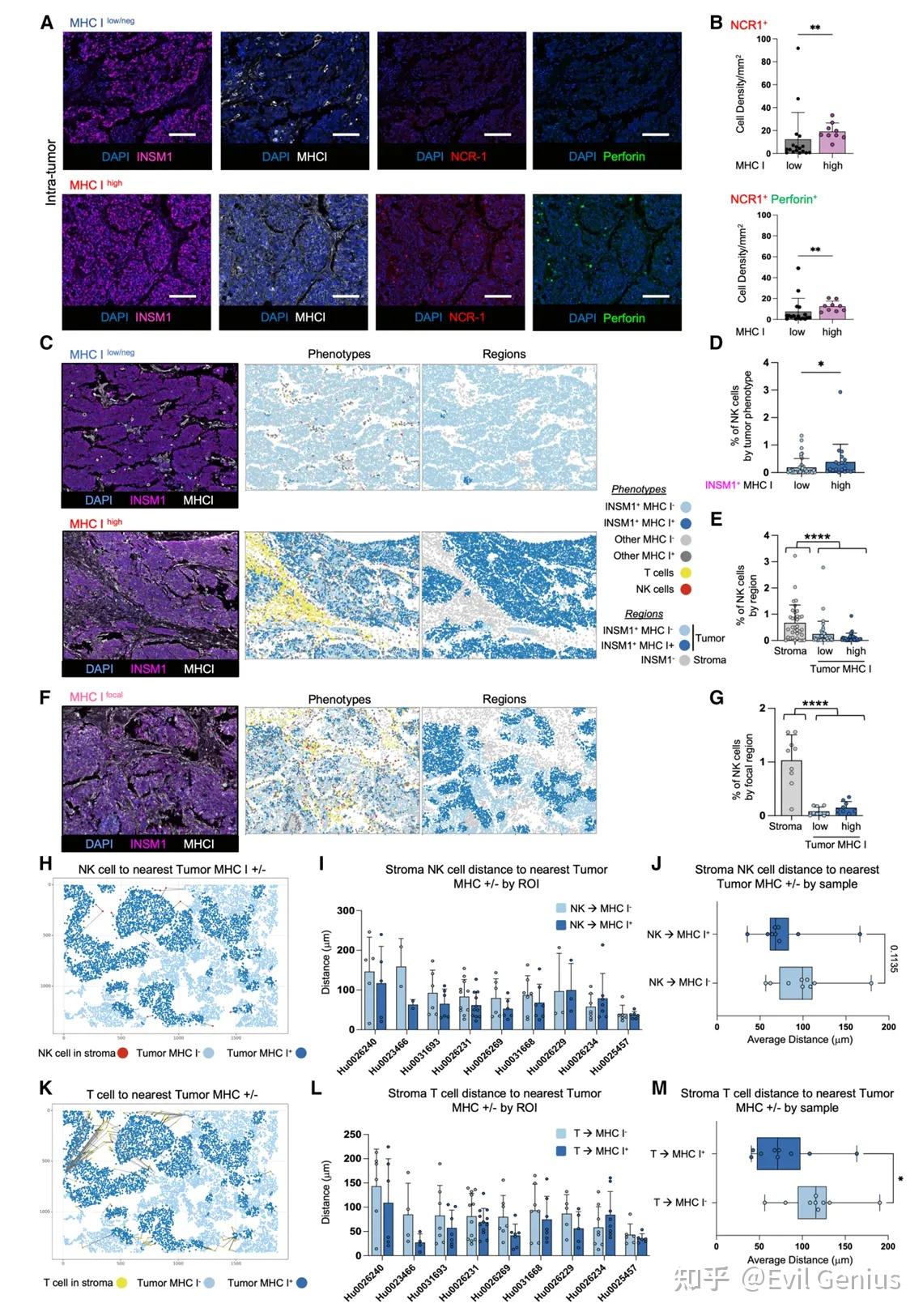
结果2、小细胞肺癌肿瘤免疫微环境的定量空间分析
免疫细胞分布与MHC-I表达相关:
在MHC-I低表达/阴性的SCLC肿瘤中,无论是肿瘤内部还是肿瘤-基质界面(TSI),NK细胞(以及T细胞)都极为稀少。
相比之下,MHC-I高表达的SCLC肿瘤中,NK细胞和T细胞的浸润显著增加,但这些免疫细胞主要分布在基质区域,而非直接进入肿瘤核心。
功能状态与空间定位:
浸润的NK细胞和T细胞异质性表达颗粒酶(Perforin),表明它们具备潜在的细胞毒性功能。
在具有局灶性MHC-I表达的肿瘤中,免疫细胞(NK和T细胞)在空间上更倾向于靠近那些高表达MHC-I的肿瘤细胞,而远离MHC-I低表达的肿瘤细胞。
临床数据验证:
通过独立临床队列的转录组数据验证,发现高MHC基因表达的SCLC患者具有更高的NK细胞特征评分。
在MHC-I低表达组中,NK细胞评分最低的患者总生存期(OS)更差。
尽管NE MHC-I低表达/阴性SCLC在体外对NK细胞杀伤高度敏感,但患者肿瘤微环境的原位分析显示,这类肿瘤的TIME中实际上排除了NK细胞(和T细胞)。免疫细胞(包括NK细胞)被限制在基质中,并倾向于靠近高表达MHC-I的肿瘤细胞,这导致了一种免疫忽视状态,解释了为何内在敏感性与临床实际免疫浸润不足之间的矛盾。
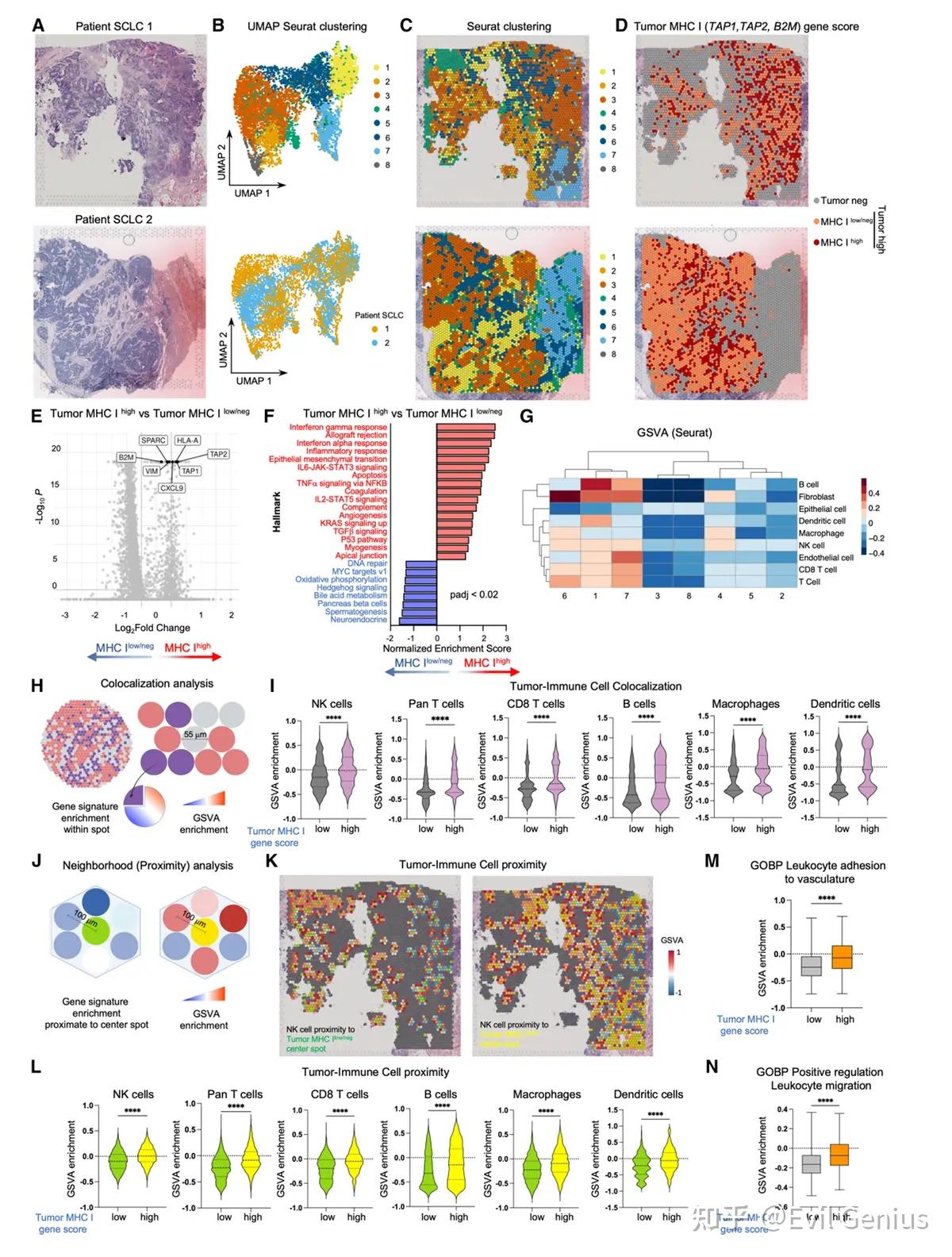
visium分析发现,NK细胞和T细胞特征与MHC-I高表达SCLC spots共定位。邻域分析显示,MHC-I高表达SCLC spots显著富集于邻近(155 μm范围内)多个免疫细胞特征(包括NK细胞)富集的spots周围。
NK细胞趋化因子失配可能解释SCLC中NK细胞运输功能障碍,同时还提示肿瘤血管系统可能存在的额外影响。
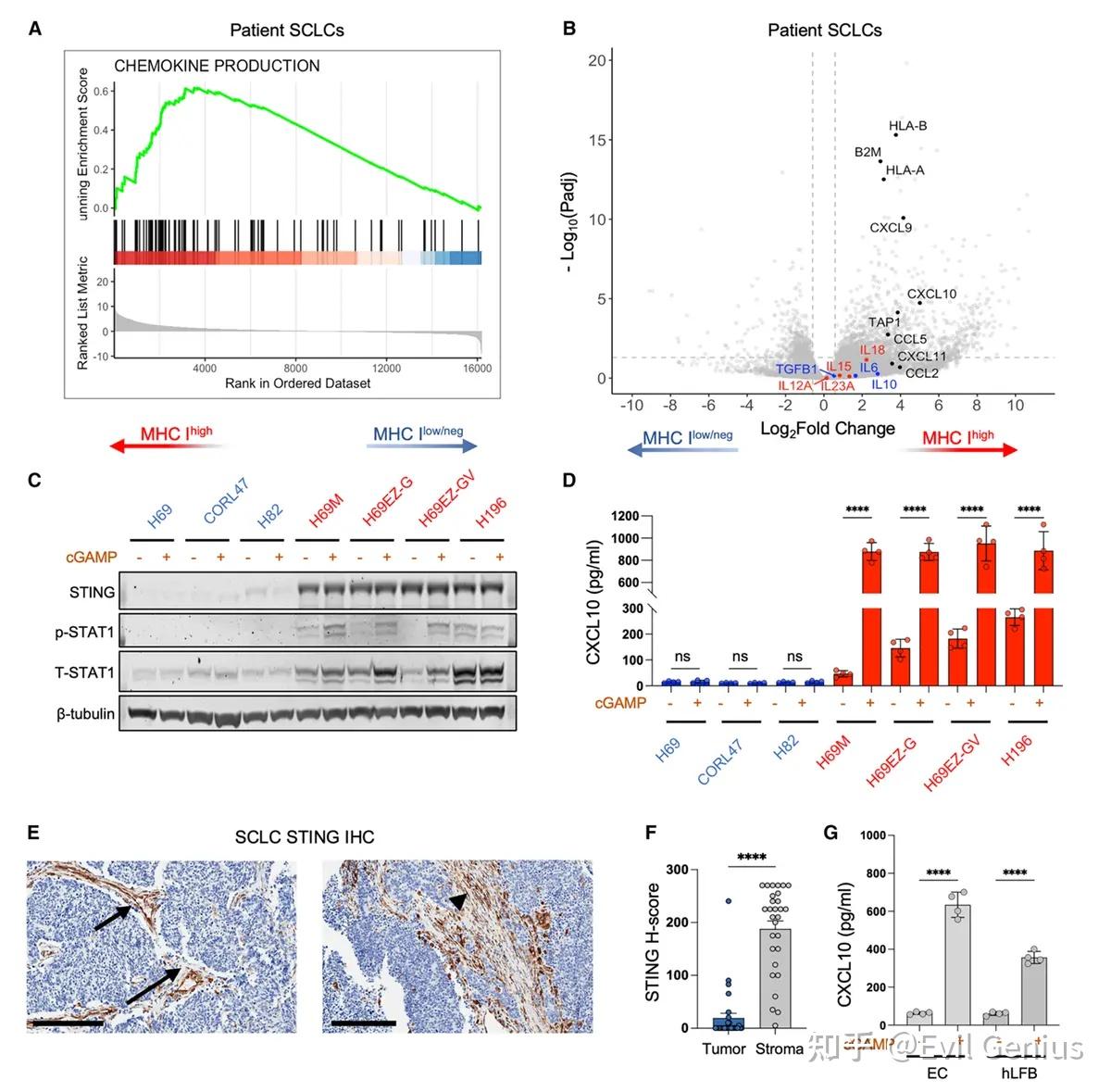
结果3、神经内分泌MHC-I阴性/弱阳性小细胞肺癌表现出免疫惰性表型
趋化因子表达缺陷:
通过患者标本的bulk RNAseq数据分析证实,与非NE MHC-I高表达SCLC相比,NE MHC-I低表达/阴性SCLC中NK细胞趋化因子(如CXCL9、CXCL10)的基因表达显著下调。
STING信号通路缺陷:
NE MHC-I低表达/阴性SCLC细胞本身缺乏STING表达。
因此,用STING激动剂(2'3'-cGAMP)处理时,这类肿瘤细胞无法有效分泌STING诱导的细胞因子/趋化因子,而非NE MHC-I高表达细胞则有强烈反应。
肿瘤微环境中的STING表达差异:
在患者肿瘤组织中,虽然肿瘤细胞(尤其是NE型)STING表达水平低,但微环境中的非恶性基质细胞(包括形态学上可识别的内皮细胞和成纤维细胞)却高表达STING。
这些基质细胞对STING激动剂有强烈的反应能力。
结果4、在小细胞肺癌肿瘤免疫微环境的三维离体模型中模拟血管-免疫细胞相互作用
研究者利用自主研发的3D微生理免疫肿瘤环境(MITE)模型,模拟并解析SCLC肿瘤微环境中血管-免疫细胞互作的过程
模型构建与实验设计
构建了包含微血管网络(HUVEC+HLF)和CORL47(MHC-I低表达/阴性SCLC)球状体的3D共培养系统,模拟肿瘤-血管-免疫界面。
通过灌注原代NK细胞(±单核细胞),并结合STING激动剂(2'3'-cGAMP)处理,探究血管和髓系细胞在免疫激活中的作用。
STING激动剂诱导炎症反应
2'3'-cGAMP处理显著诱导炎症细胞因子/趋化因子产生,尤其在存在单核细胞时效果最强。
CXCL10(关键NK细胞趋化因子) 的表达在cGAMP+单核细胞条件下显著升高。
DynaMITE-seq揭示细胞特异性响应
单细胞测序鉴定出5种细胞类型(EC、成纤维细胞、SCLC、NK细胞、单核细胞)。
转录响应具有细胞类型特异性:
非恶性细胞(单核细胞、EC、成纤维细胞) 在cGAMP处理后显著上调STING介导的干扰素信号通路及趋化因子(CXCL10/CXCL11)。
CORL47 SCLC细胞则无此响应,印证其STING功能缺陷。
CXCL10主要来源于cGAMP刺激后的非恶性细胞(尤其是单核细胞和EC)。
NK细胞功能状态优化
在cGAMP+单核细胞条件下,NK细胞呈现激活表型:
上调:激活受体/标志物(FCER1G、CD69)、细胞毒性基因(GZMB、TNFSF10)。
下调:抑制性受体(KLRD1、TIM3)、趋化受体CXCR4。
模型生理相关性验证
MITE各细胞类型与正常人类肺组织单细胞图谱高度对应。
SCLC细胞cluster与肺神经内分泌细胞(SCLC潜在祖细胞)存在转录相似性。
MITE数据与患者SCLC空间转录组数据良好匹配,验证了模型的临床相关性。
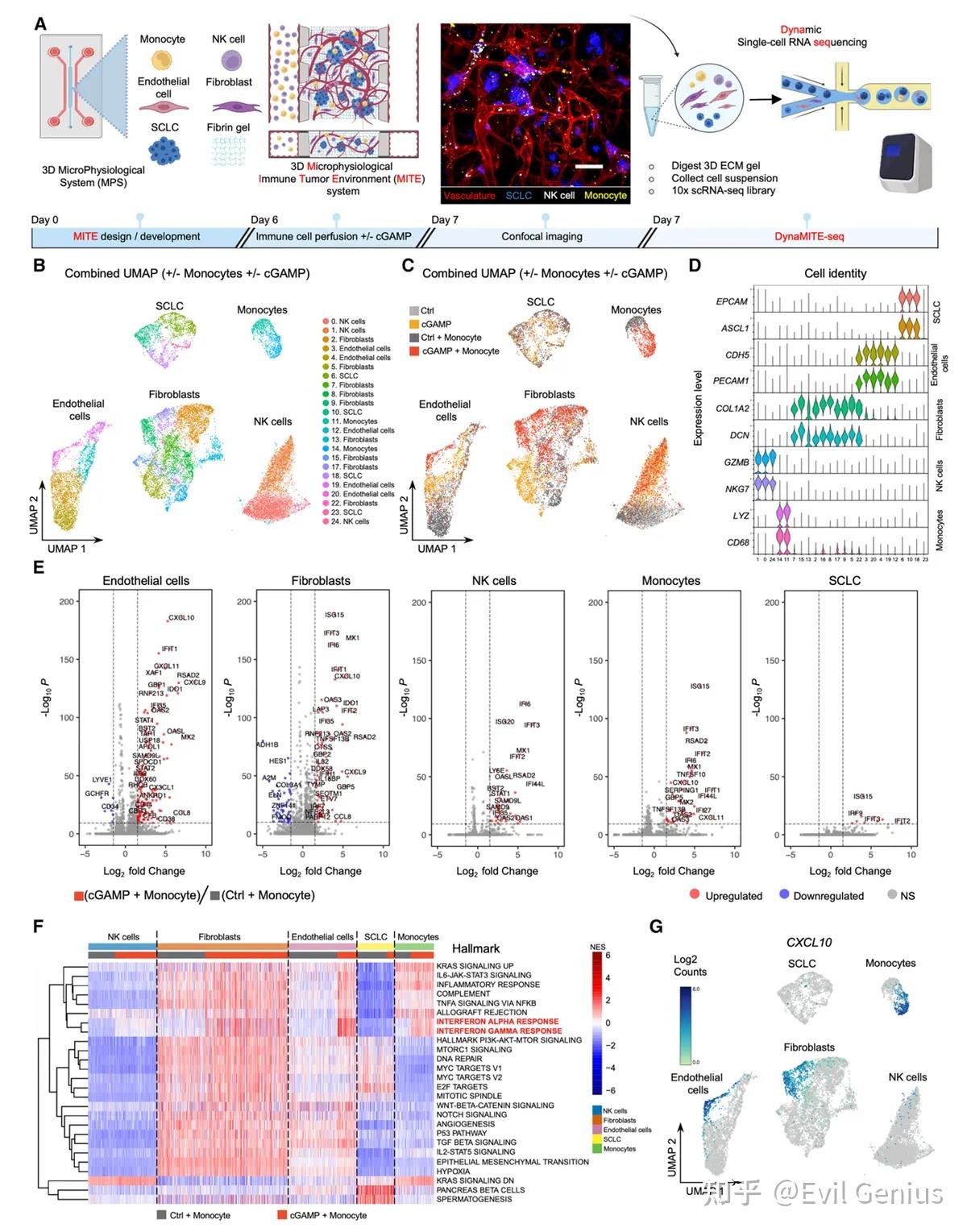
通过CellChatDB算法分析了STING激动剂(2'3'-cGAMP)处理对MITE模型中细胞间通讯的影响,尤其是血管内皮细胞与NK细胞的互作
STING激动剂增强细胞间通讯
2'3'-cGAMP刺激显著增加了MITE中细胞间相互作用的数量和强度。
在含有单核细胞的条件下,cGAMP处理诱导了白细胞运输(CCL家族) 和血管外渗(SELE、VCAM) 相关通路上调。
内皮细胞-NK细胞互作增强
RLI配对分析显示,cGAMP+单核细胞条件下,内皮细胞(EC)来源的SELE和VCAM1与NK细胞的相互作用显著增强。
网络分析进一步证实,EC通过VCAM和SELE信号通路与NK细胞建立通讯,且这些EC主要富集于cGAMP+单核细胞处理组。
其他通路分析还提示,cGAMP促进EC-NK细胞黏附(ICAM1)、单核细胞-NK细胞趋化(CCL家族) 以及NK-SCLC黏附(CD96)。
内皮细胞亚群分析揭示激活表型
对EC进行亚聚类分析发现,cGAMP+单核细胞条件下,EC富集了炎症反应和白细胞外渗相关通路,包括CXC家族趋化因子。
原代肺内皮细胞验证
原代人肺微血管内皮细胞(LEC) 经STING激动剂(cGAMP或ADU-S100)处理后,CXCL10分泌增加,同时SELE、ICAM1、VCAM1转录上调。
敲低EC中的STING可完全阻断上述分子的上调,证实STING信号在血管激活表型中的关键作用。
核心结论:
通过CellChat分析和体外验证,研究者证明:STING激动剂可激活肿瘤微环境中的血管内皮细胞,诱导其表达黏附分子(SELE、VCAM1、ICAM1)和趋化因子(CXCL10等),从而促进NK细胞的招募、黏附和外渗。这揭示了STING激动剂通过重塑血管表型来克服SCLC免疫排斥的潜在机制,为改善NK细胞疗法提供了新策略。
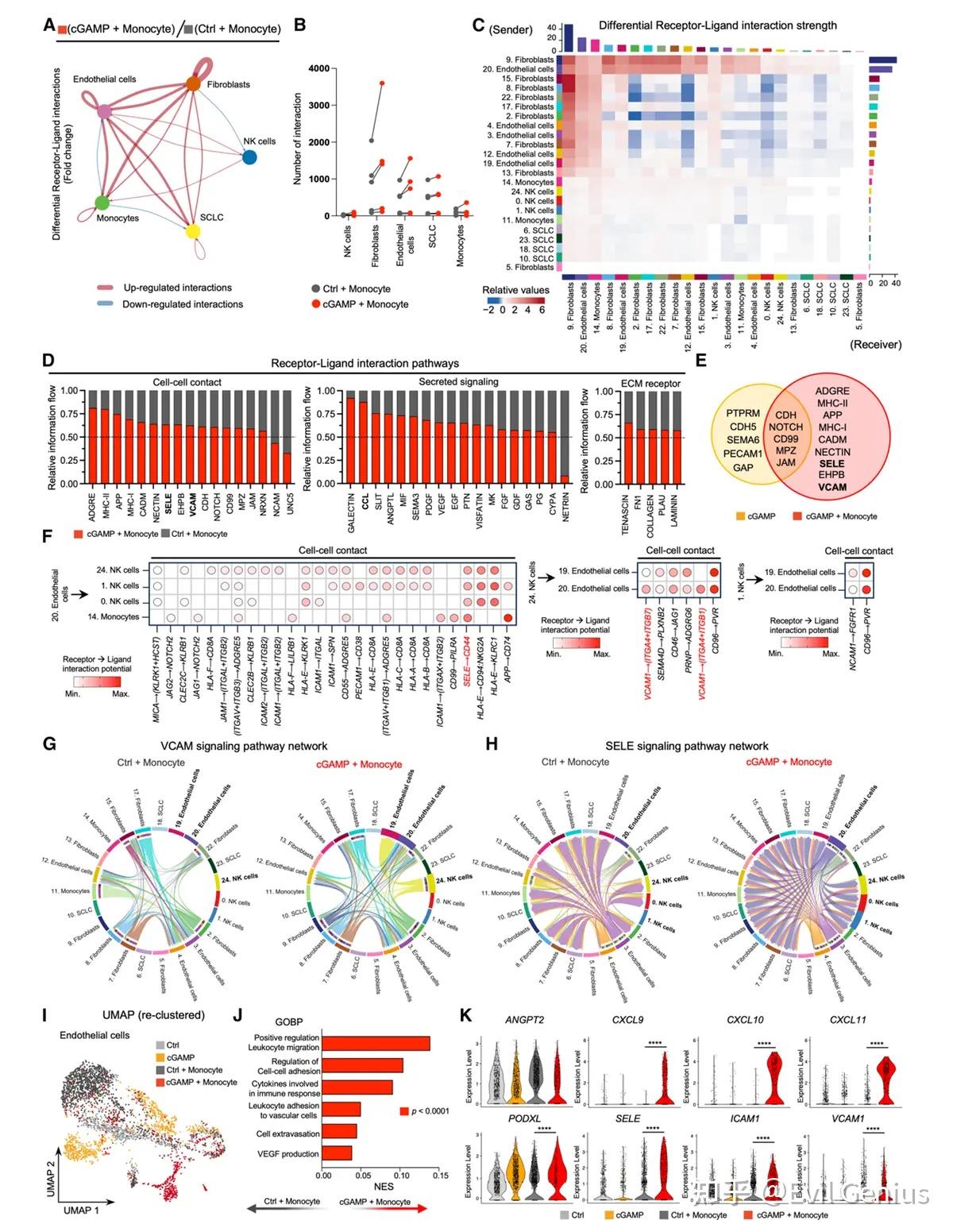
结果5、干扰素基因刺激因子介导的血管激活在小细胞肺癌模型中的原位与体内验证
通过STING预激活血管,可以增强治疗性NK细胞在NE MHC-I低表达/阴性SCLC中的募集。
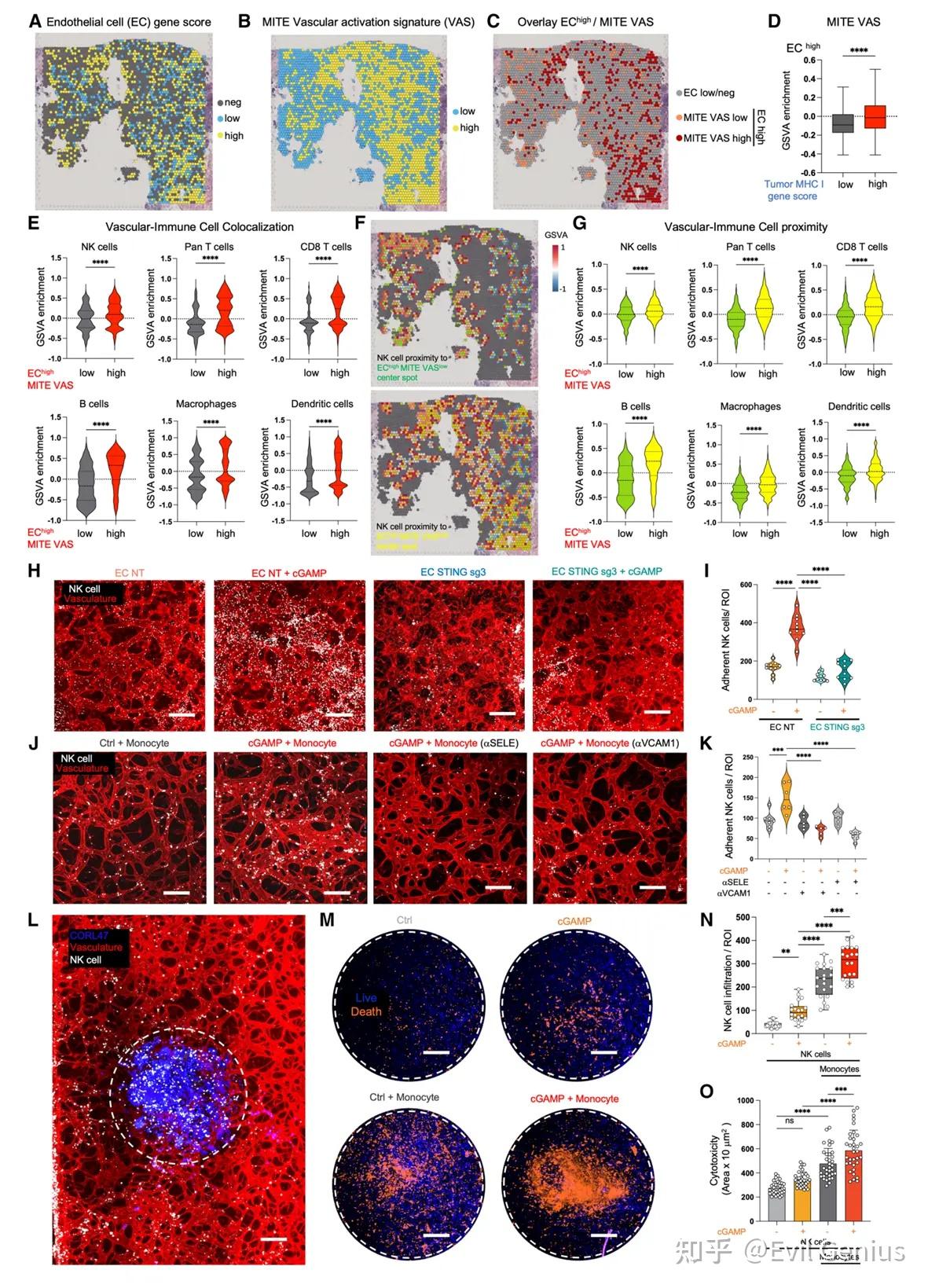
结果6、干扰素基因刺激剂介导的血管激活可增强NK细胞迁移并靶向小细胞肺癌
STING激动剂增强NK细胞血管外渗和杀伤功能(体外3D模型验证)
血管屏障功能调控:
在宏观血管模型中,2'3'-cGAMP以STING依赖性方式增加血管屏障对多种尺寸葡聚糖的通透性,提示STING激动可能增强血管被动扩散。
NK细胞黏附机制:
在微血管网络(MVN)模型中,2'3'-cGAMP刺激显著增强NK细胞对血管壁的黏附,且在单核细胞存在时效果最强。
敲除EC中的STING或抗体阻断SELE/VCAM1均可消除这一效应,证实EC-STING信号及下游黏附分子是NK细胞 Trafficking 的关键。
抗肿瘤功能验证:
在单球体-MITE(ms-MITE)模型中,2'3'-cGAMP+单核细胞处理显著增强NK细胞对MHC-I低表达/阴性SCLC的浸润和杀伤能力。
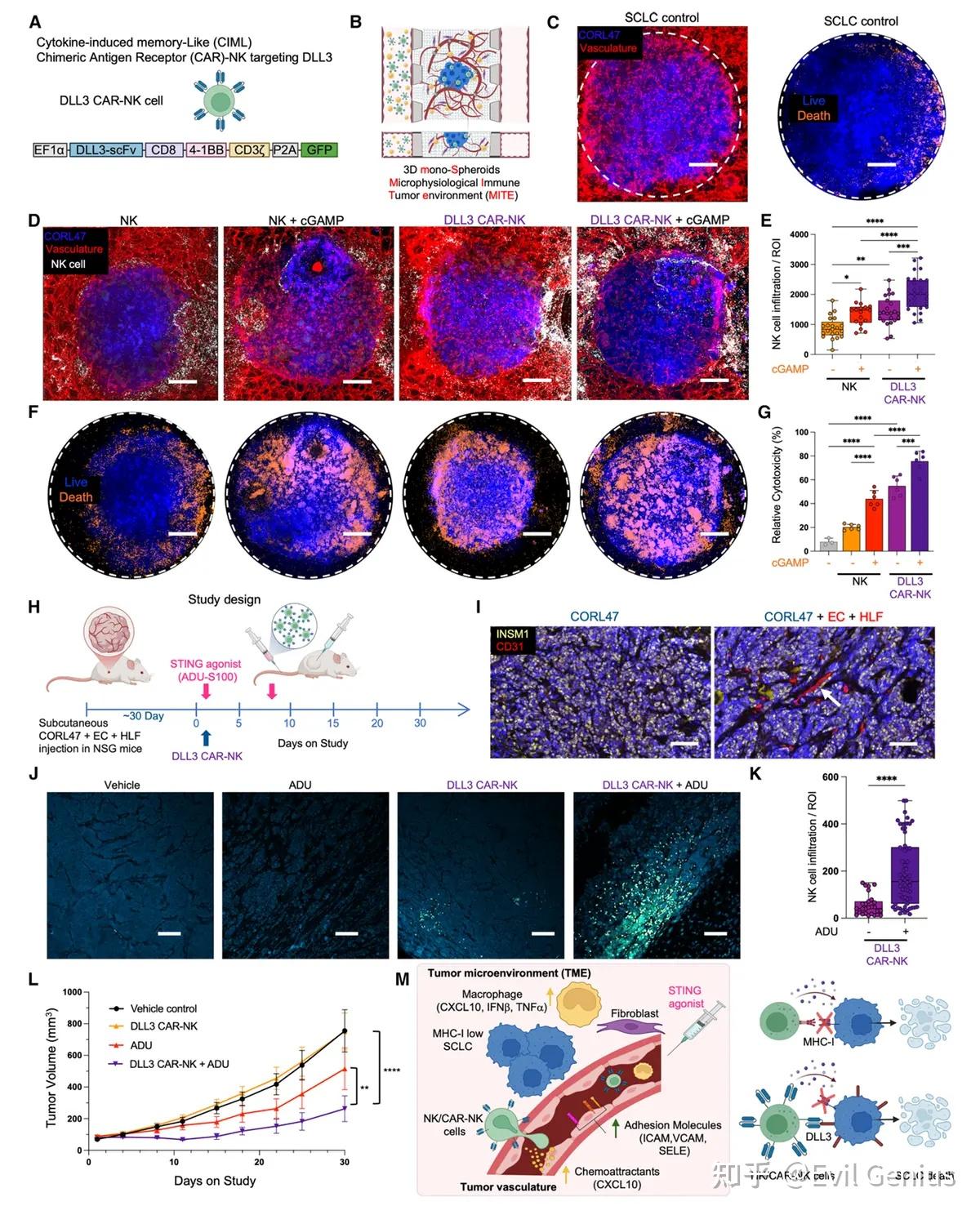
STING激动剂联合免疫治疗策略探索
联合PD-1阻断(原位NK细胞):
在同源小鼠模型(RPR2)中,系统性STING激动剂(dazostinag)+ anti-PD-1联合治疗虽仅显示抗肿瘤趋势,但显著增加了肿瘤内NK细胞和T细胞浸润。
同时耗竭NK细胞和CD8+ T细胞可显著削弱联合治疗疗效,表明NK细胞在该联合策略中发挥重要作用,但内源性NK细胞数量稀少可能限制其疗效。
联合CAR-NK细胞治疗(过继性转移):
构建DLL3靶向的CAR-NK细胞,其在体外对CORL47细胞(包括B7-H6缺陷株)显示增强的杀伤能力。
在MITE模型中,DLL3 CAR-NK细胞联合2'3'-cGAMP显著增强血管黏附和抗肿瘤细胞毒性。
体内验证(人源化血管SCLC异种移植模型)
在建立人源化血管的CORL47肿瘤模型中,瘤内注射ADU-S100 + 静脉输注DLL3 CAR-NK细胞联合治疗组:
显著增加肿瘤内CAR-NK细胞浸润。
显著抑制肿瘤生长,效果优于任一单药治疗。
核心结论:
本研究通过系列功能实验证明:STING激动剂通过激活血管内皮细胞(上调SELE/VCAM1等黏附分子),促进NK细胞(包括CAR-NK)的血管外渗和肿瘤浸润。在此基础上,STING激动剂联合DLL3 CAR-NK细胞治疗在SCLC模型中显示出协同抗肿瘤效应,为克服SCLC免疫排斥、提高过继性NK细胞疗法的疗效提供了理论依据和转化策略。
最后来看看方法


生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号