Nat Commun | AI 药物发现的下半场:为什么 Roche 坚持用 13490 个实验负结果来校准反应预测模型?

Nat Commun | AI 药物发现的下半场:为什么 Roche 坚持用 13490 个实验负结果来校准反应预测模型?

MindDance
发布于 2026-01-22 12:34:12
发布于 2026-01-22 12:34:12
在药物研发的漫长征途中,从苗头化合物(Hit)到先导化合物(Lead)的优化阶段往往是效率的瓶颈。如何快速、经济地合成并筛选出既具有高活性又具备优良药理特性的候选分子,一直是药物化学家的核心挑战。近期,《Nature Communications》发表了一项由 Roche 研究团队主导的创新研究。该研究展示了一个整合了微型化高通量实验(HTE)、深度学习反应预测以及多维度属性优化(MDO)的药物化学工作流。通过这一流程,研究团队针对单酰基甘油脂肪酶(MAGL)这一重要靶点,成功将命中化合物的活性提升了高达 4500 倍,并实现了亚纳摩尔级的抑制效力。这一成果不仅为 MAGL 抑制剂的开发提供了新思路,更展示了AI 与自动化实验深度融合在加速药物发现进程中的巨大潜力。
背景:药物化学中的“合成-筛选”困局
在传统的药物化学优化过程中,研究人员通常依赖于迭代诱变和点突变策略来提升分子的活性。然而,这种方法往往受限于合成的可及性和有限的序列/结构空间探索。尤其是对于复杂的杂环骨架,实现位点特异性的功能化往往需要繁琐的多步合成,这极大地限制了先导化合物优化的速度。
单酰基甘油脂肪酶(MAGL)是内源性大麻素系统中的关键酶,与神经炎症和多种中枢神经系统疾病密切相关,是极具吸引力的药物靶点。尽管已有部分 MAGL 抑制剂进入临床研究,但开发具有高度特异性、优良药代动力学(ADMET)属性且合成简便的新型抑制剂仍是当务之急。为了突破这一困局,Roche 团队探索了利用Minisci 型 C-H 烷基化反应进行后期功能化的可能性,并试图通过计算手段预判反应结果和分子属性。
方法:AI 驱动的自动化优化漏斗
研究团队构建了一个精密的设计-合成-测试闭环,其核心在于将实验产生的高质量数据与先进的机器学习模型相结合。
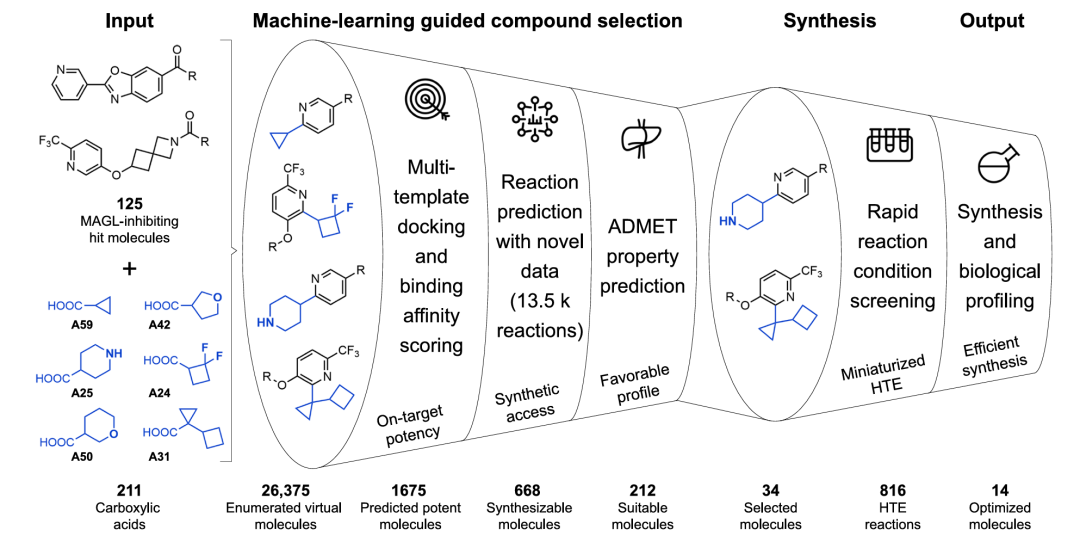
命中化合物优化工作流,展示从虚拟库枚举到最终优化分子的多级筛选过程
命中化合物优化工作流,展示从虚拟库枚举到最终优化分子的多级筛选过程
- 高质量反应数据集的生成: 利用微型化高通量实验,研究团队针对 Minisci 反应生成了一个包含 13,490 个新颖反应的数据集。这一数据集涵盖了 80 个含氮杂环片段和 59 个羧酸片段的多种组合。通过液相色谱-质谱(LC-MS)分析,研究人员获得了精确的反应产率数据,为后续模型训练奠定了坚实基础。
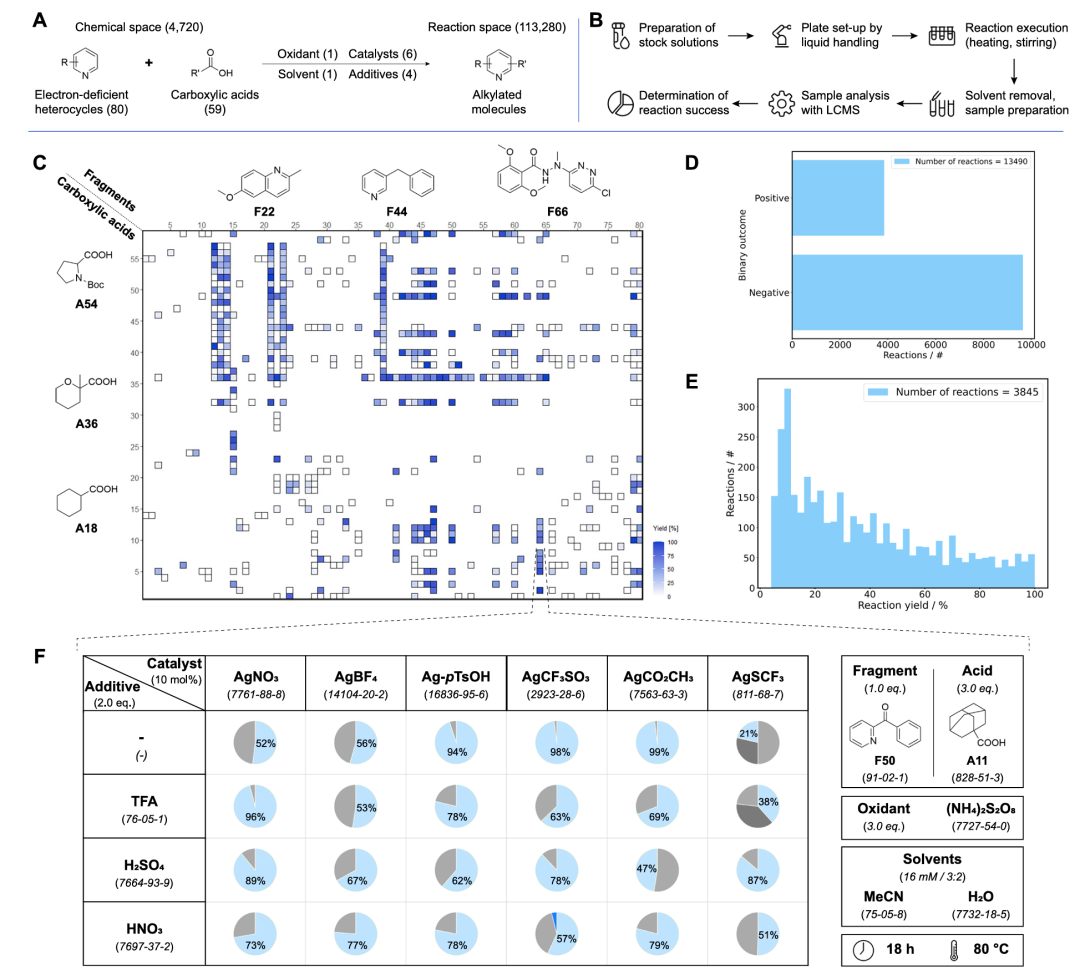
HTE 反应筛选矩阵与产率分布图
HTE 反应筛选矩阵与产率分布图
- 深度学习反应预测模型: 基于上述数据集,研究团队训练了深度图神经网络。该模型能够准确预测反应的成功率(二元分类)和产率(回归分析)。在随机划分的数据集上,模型表现出极高的预测精度(准确率达 85.7%),甚至在面对未见过的酸或杂环片段时,仍保持了良好的泛化能力。
- 多维度属性优化(MDO)策略: 研究团队枚举了一个包含 26,375 个潜在 Minisci 反应产物的虚拟化学库。随后,利用一个多级“过滤漏斗”进行筛选:
- 活性预测:利用多模板分子对接和结合亲和力评分,筛选出潜在的高活性分子。
- 合成可及性评估:利用训练好的 GNN 模型预测反应产率,确保候选分子能够被高效合成。
- ADMET 属性预测:评估分子的亲脂性(LogD)、溶解度(LYSA)、渗透性(PAMPA)等关键药理参数。
这种 MDO 策略避免了单一指标的顾此失彼,确保了最终选定的分子在活性和成药性之间达到最佳平衡。
结果:从亚微摩尔到亚纳摩尔的飞跃
通过这一工作流,研究团队从 212 个候选分子中精选并合成了 14 个化合物。实验结果令人振奋:
- 效力的显著提升: 所有新合成的化合物均表现出增强的抑制活性。其中,化合物 17 的 IC50 达到亚纳摩尔级别,相比起始的苗头化合物提升了 4500 倍。
- 优异的药理特征: 选定的化合物不仅活性高,还表现出良好的亲脂效率(LipE)和跨膜渗透性。更重要的是,这些分子对 MAGL 表现出极高的选择性,避免了对其他脑部水解酶的脱靶作用。
- 结构生物学验证: 研究团队获得了三个计算设计配体与 MAGL 蛋白的共晶结构。结构分析证实,通过 Minisci 反应引入的环戊基或环丙基-环丁基基团完美契合了酶活性位点的一个此前未被利用的疏水亚口袋,并诱导了吡啶环的翻转,形成了新的氢键网络。
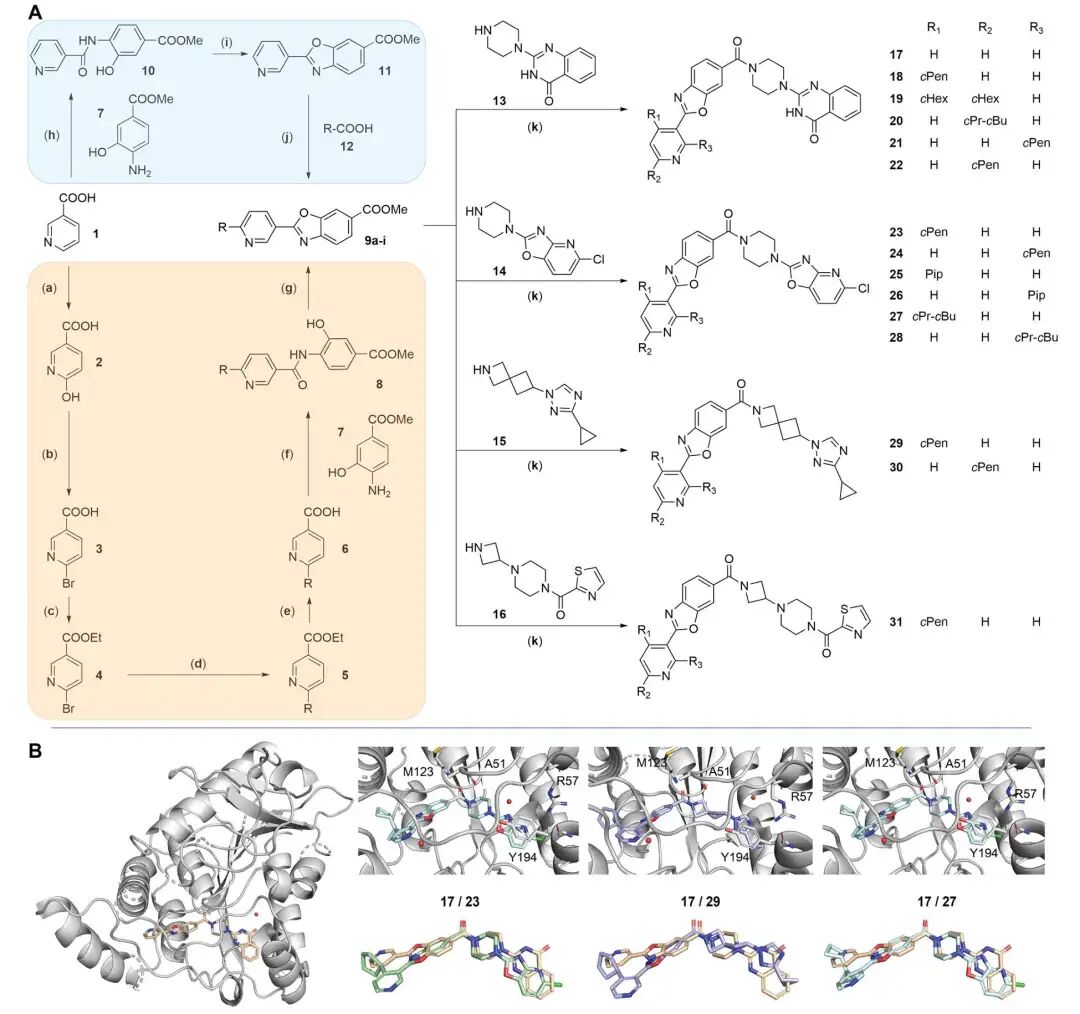
共晶结构分析,展示设计分子与靶点的相互作用模式
共晶结构分析,展示设计分子与靶点的相互作用模式
思考
这篇论文不仅是一次成功的药物优化实践,更在方法论层面为我们带来了深层剖析与启示:
- 数据质量胜过算法复杂度: 该研究成功的基石并非某种极其复杂的黑盒算法,而是通过 HTE 获得的 13,490 个高质量、标准化且包含负结果的实验数据。在 AI 药物发现领域,“垃圾进,垃圾出”是永恒的真理。Roche 的工作再次证明,拥有自主产生高质量实验数据的能力,是药企在 AI 时代的核心竞争力。
- 后期功能化(LSF)与 AI 的完美契合: Minisci 反应等 C-H 活化反应虽然强大,但其区域选择性和底物兼容性往往难以预测。AI 恰好擅长处理这种复杂的非线性映射。将 AI 反应预测应用于 LSF,可以极大地扩展药物化学家的“合成工具箱”,实现对复杂分子的“外科手术式”修饰。
- 从“单点突破”到“系统优化”: 很多 AI 设计的研究只关注活性预测,而忽略了合成难度和 ADMET 属性。本研究展示的 MDO 漏斗提供了一个更具实战意义的范式:在设计的早期阶段就引入合成可及性和药理约束。这种“以终为始”的设计思路,能够显著降低后期开发的失败率。
- 物理模型与机器学习的协同: 研究中结合了分子对接(物理/几何约束)与 GNN(数据驱动预测)。这种混合驱动的模式,既利用了物理规律的严谨性,又发挥了机器学习处理复杂模式的灵活性,是当前计算药学的主流趋势。
值得思考的问题
Q: 在反应预测模型中,包含“负结果”(即产率极低或失败的反应)数据有多重要?
A: 至关重要。大多数公开数据库(如 USPTO)主要记录成功的反应,这会导致模型产生严重的“幸存者偏差”。本研究通过 HTE 产生了约 70% 的负结果数据,这使得模型能够准确识别哪些反应行不通,从而在虚拟筛选中有效排除无法合成的分子,极大地提升了筛选的实战价值。
Q: 为什么选择 Minisci 反应作为优化的核心工具,而不是传统的偶联反应?
A: Minisci 反应属于后期功能化(LSF)技术,它允许在不预先功能化底物的情况下,直接在复杂的杂环骨架上引入烷基。这对于先导化合物优化阶段的快速多样化非常有利。相比之下,传统的偶联反应通常需要预先合成带卤素或硼酸基团的中间体,步骤繁琐且灵活性较低。
Q: 该工作流中的多维度属性优化(MDO)是如何避免模型误差累积的?
A: 研究团队采用了非固定阈值的顺序过滤策略。在筛选过程中,并不简单地对每个指标设置死板的截断值,而是结合了预测的置信度和多指标的综合评分。这种方法允许在某一指标稍弱但在其他指标表现优异的分子进入下一轮,从而在一定程度上缓解了单个模型预测误差对整体结果的干扰。
Q: 这种基于特定反应(如 Minisci)的优化策略,是否会限制化学空间的探索?
A: 确实存在这种权衡。虽然限制在特定反应内会缩小理论上的化学空间,但它换取了极高的合成成功率。在药物研发的先导优化阶段,“能做出来的分子”远比“理论上完美的分子”更有价值。此外,随着更多反应类型的 HTE 数据被整合进工作流,可探索的化学空间将不断扩大。
参考文献:Nippa, D.F., Atz, K., Stenzhorn, Y. et al. Expediting hit-to-lead progression in drug discovery through reaction prediction and multi-dimensional optimization. Nat Commun 16, 11646 (2025). https://doi.org/10.1038/s41467-025-66324-4
代码数据:
https://github.com/ETHmodlab/minisci
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-01-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号