当AI学会「搭积木」:这款新型算法为多目标药物设计开辟新路径

当AI学会「搭积木」:这款新型算法为多目标药物设计开辟新路径

MindDance
发布于 2026-01-08 12:53:37
发布于 2026-01-08 12:53:37
💊 药物设计的世纪难题:如何同时满足多个「理想属性」?
在药物研发的浩瀚宇宙中,科学家们面临着一个核心挑战:如何设计出同时具备多种理想特性的分子——比如高效结合靶点、低毒性、高生物利用度等。传统方法如同在10⁶⁰种可能的分子空间中「大海捞针」,而多目标药物设计更像是在走钢丝:每增加一个优化目标,难度便呈指数级上升。
近年来,AI驱动的分子生成模型为这一领域带来曙光,但 Graph-based 方法仍存在三大瓶颈:
- 1. 化学空间与有效性的矛盾:片段法生成的分子虽合规却缺乏多样性,原子法虽灵活却易产生无效结构;
- 2. 数据稀缺性:实验测量的分子属性数据有限,制约模型预测精度;
- 3. 任务适应性差:传统模型参数高度依赖特定任务,难以快速迁移至新需求。
✨ ScafVAE:用「键骨架」搭出药物分子的「AI设计师」
中山大学与北京大学团队联合开发的 ScafVAE(骨架感知变分自动编码器),通过三大创新设计突破了上述瓶颈:
1. 键骨架生成:在「搭积木」中平衡创新与合规
不同于传统的「原子逐个拼接」或「预定义片段组装」,ScafVAE提出「键骨架」概念:先构建仅包含键类型的「分子骨架」,再逐步装饰原子类型。这种方法如同先确定积木的连接方式,再填充具体模块,既保留了片段法的化学有效性,又通过数据驱动的碎片化策略(基于「键困惑度」指标)扩展了化学空间。实验表明,该策略使生成分子的有效性达0.987,且在GuacaMol基准测试中,其生成分子的新颖性和多样性超越所有SMILES-based模型。
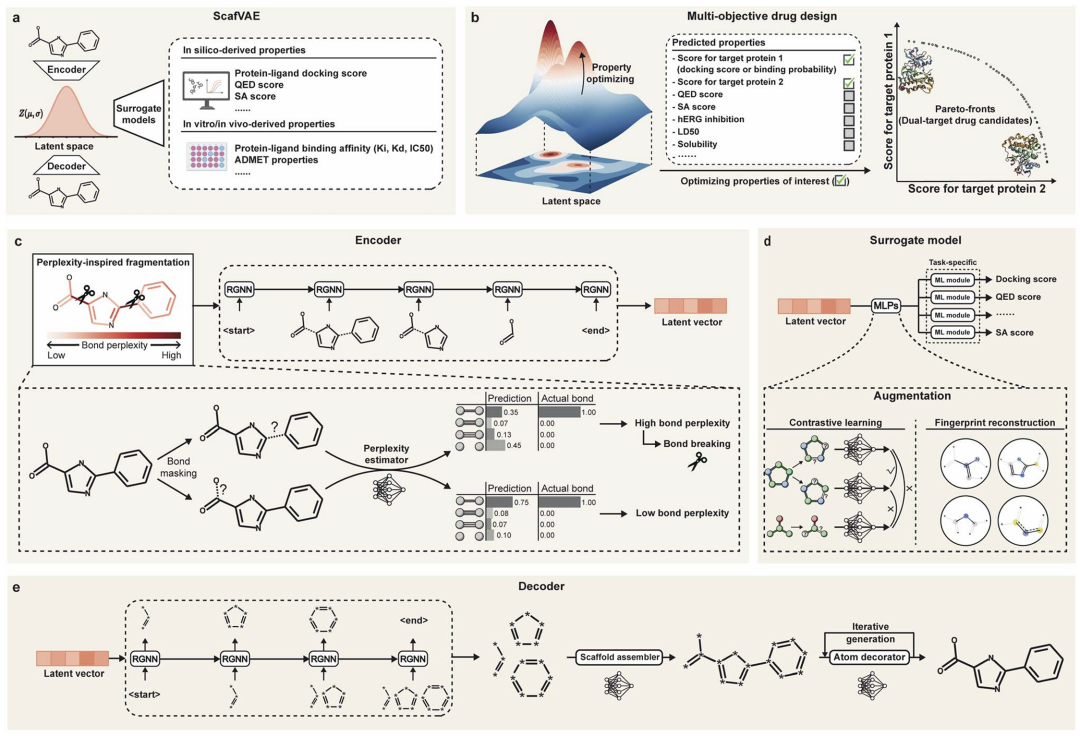
图1:ScafVAE的解码器首先生成键骨架,再通过原子装饰迭代生成最终分子。
图1:ScafVAE的解码器首先生成键骨架,再通过原子装饰迭代生成最终分子。
ScafVAE摒弃了传统的原子或片段生成方法,独创了一种“键骨架为本”(bond scaffold-based)的生成策略 。它首先生成一个仅包含化学键类型和连接方式的“骨架”(bond scaffold),然后再用合适的原子类型去“装饰”这个骨架,最终形成一个完整的、化学性质稳定的分子。这一设计巧妙地融合了原子生成法和片段生成法的优点,既保留了后者生成分子的化学高合理性,又极大地拓展了可探索的化学空间,让发现新颖结构成为可能。
2. 困惑度启发碎片化:让AI学会「聪明拆分子」
ScafVAE引入「键困惑度」作为碎片化指导原则,通过预训练的图模型估算每条键的不确定性,优先断裂高困惑度的键。相比传统规则化方法(如BRICS),该方法可100%成功拆解分子,且生成的片段连接点更多,词汇量减少1-2个数量级,大幅提升模型效率。
3. 代理模型增强:用「预训练+对比学习」攻克数据稀缺
通过对比学习和分子指纹重建,ScafVAE的代理模型仅需少量任务特定参数,即可高效预测20种ADMET性质(如毒性、代谢稳定性等)。在ADMET任务中,其Spearman's ρ值达0.73,较JT-VAE提升6%,且单样本预测时间仅1.82×10⁻⁴秒,几乎可忽略不计。
🚀 实战验证:从双靶点抗癌药到多属性优化
🔬 双靶点药物设计:对抗癌症耐药性的「组合拳」
研究团队针对癌症治疗中的四大耐药机制(表观遗传改变、药物外排、DNA修复、细胞死亡失调),利用ScafVAE设计双靶点药物。例如,在EGFR/HER2靶点组合中,生成分子的实际对接分数低至-13.3 kcal/mol,且通过分子动力学(MD)模拟验证了其与靶点的稳定结合。更令人振奋的是,结合实验测量的结合亲和力数据时,生成分子的结合概率超0.95。
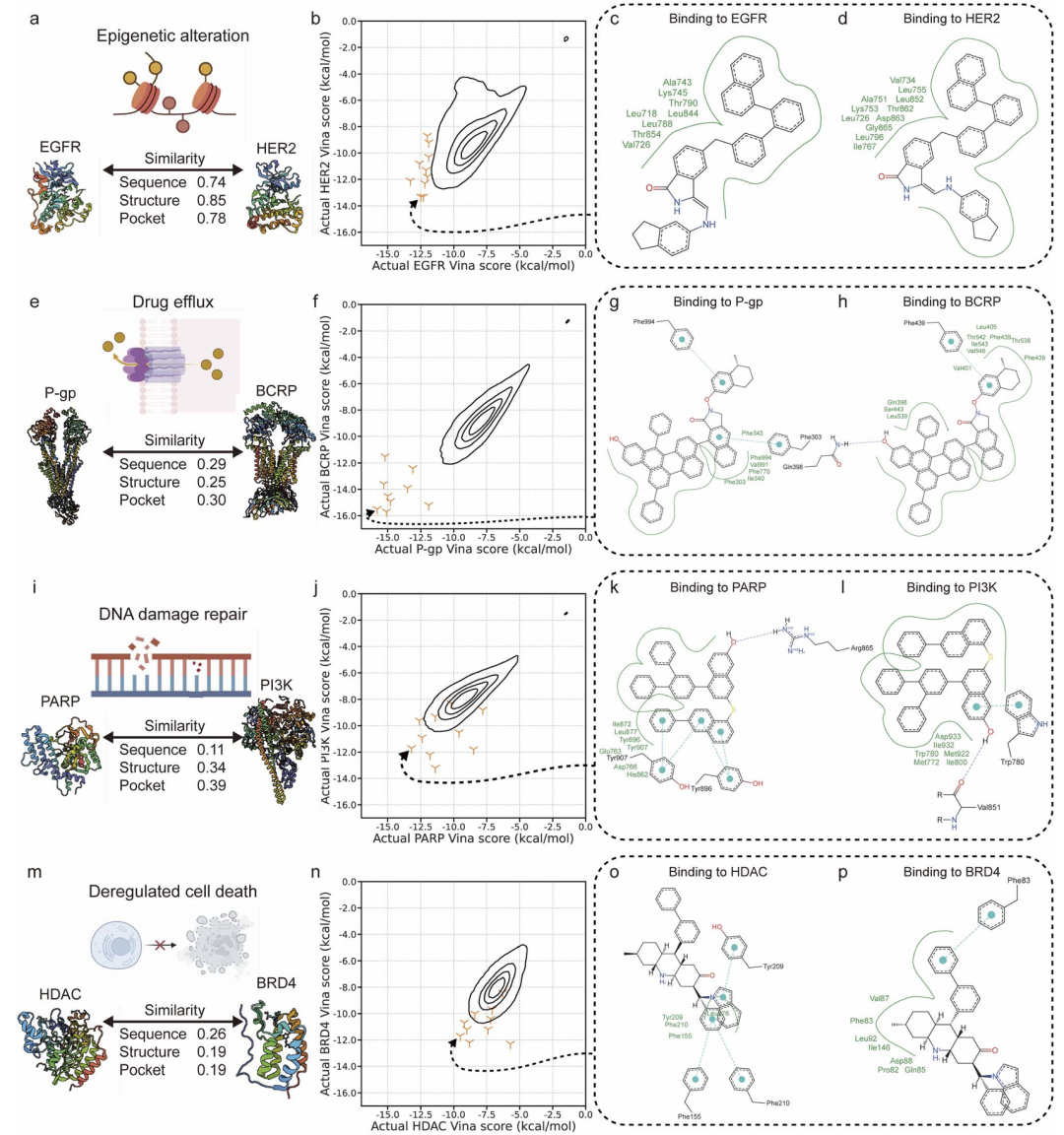
图2:ScafVAE针对四种耐药机制生成的双靶点分子及其与靶蛋白的相互作用。
图2:ScafVAE针对四种耐药机制生成的双靶点分子及其与靶蛋白的相互作用。
🌈 多目标优化:一次满足「药效+成药性」
当纳入QED(药物相似性)、SA(合成可及性)等额外属性时,ScafVAE通过NSGA-II算法实现多目标平衡。以EGFR/HER2靶点为例,优化后的分子在保持强对接分数的同时,QED分数提升至0.76,且符合Lipinski「五规则」——这意味着它们更可能成为可开发的候选药物。
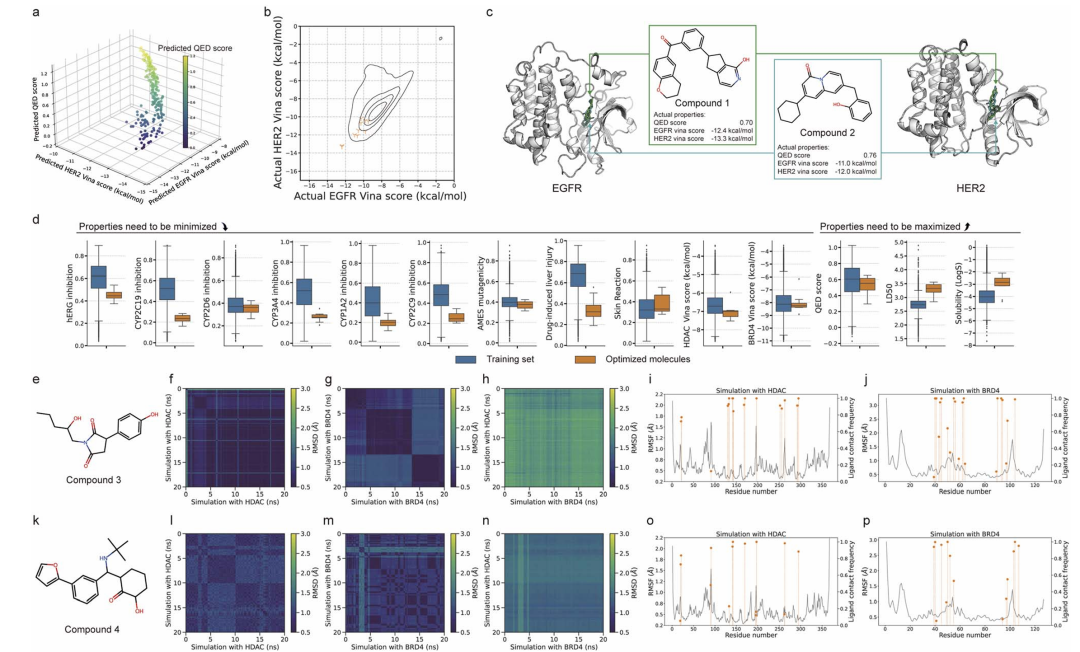
图3:多目标优化生成的分子(上)及分子动力学模拟验证其与靶蛋白的稳定结合(下)
图3:多目标优化生成的分子(上)及分子动力学模拟验证其与靶蛋白的稳定结合(下)
🔬 突破与局限:AI药物设计的「现在与未来」
ScafVAE在GuacaMol基准中展现出媲美顶尖字符串模型的性能,其图结构建模能力为复杂分子设计提供了新范式。然而,多目标冲突(如对接分数与QED的权衡)、分子几何建模缺失仍是待攻克的难题。未来,团队计划引入「一键生成」原子装饰流程,并探索键骨架与分子动力学的结合,以实现更精准的三维结构预测。
📚 论文信息
- • 标题:Multi-objective drug design with a scaffold-aware variational autoencoder
- • 期刊:Chemical Science
- • 作者:Tiejun Dong, Linlin You*, Calvin Yu-Chian Chen*
- • 链接:https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc08736d
- • 开源代码:https://github.com/tiejundong/ScafVAE
- • 发表日期:2025年6月25日
💬 结语
从「偶然发现」到「理性设计」,AI正推动药物研发进入精准时代。ScafVAE的「键骨架」智慧,或许正是打开多目标药物设计黑箱的一把钥匙——当AI学会像化学家一样理解分子骨架的奥秘,我们离「量身定制」的理想药物又近了一步。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-06-29,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号