AI+Drug 文献速递 | 大模型链式思考 × 多模态融合:刷新药物分子性质预测准确度与可解释性新高度

AI+Drug 文献速递 | 大模型链式思考 × 多模态融合:刷新药物分子性质预测准确度与可解释性新高度

MindDance
发布于 2026-01-08 12:51:37
发布于 2026-01-08 12:51:37
1. Effective And Explainable Molecular Property Prediction By Chain-Of-Thought Enabled Large Language Models And Multi-Modal Molecular Information Fusion
期刊: Journal of Chemical Information and Modeling 链接: https://doi.org/10.1021/acs.jcim.5c00577 代码: https://github.com/jinchang1223/LLM-MPP
简介: 作者提出了 LLM-MPP,一种面向药物分子性质预测的新型多模态框架。该方法同时利用 1D SMILES、2D 分子图和从 PubChem 提取的性质文本描述,通过 LoRA 微调的 LLaMA3-8b 生成链式思考(CoT)推理文本,以提升可解释性和跨模态对齐能力。模型以 AttentiveFP 提取图表示,再借助多头交叉注意力与加权融合将文本与图特征整合,并辅以温度化对比学习约束,使不同模态共享一致的潜在空间,从而获得信息丰富且鲁棒的分子表示。
在九个 MoleculeNet 基准数据集(BBBP、BACE、ClinTox、SIDER、Tox21、FreeSolv、ESOL、Lipo、QM7)的评测中,LLM-MPP 在五个数据集取得最佳成绩、一个数据集列第二,总体超越了 22 个现有模型(含 7 个 LLM 基线),其中在 BBBP 的 ROC-AUC 达 0.973,在 FreeSolv 的 RMSE 降至 1.505,验证了多模态融合和 CoT 推理对分类与回归任务的有效性。消融实验显示,冻结 LLM、去除 CoT 或文本描述都会显著降低性能,证明三模态输入、交叉注意力及对比学习设计的协同价值。作者已公开完整代码与数据处理脚本,方便社区复现实验并进一步改进。
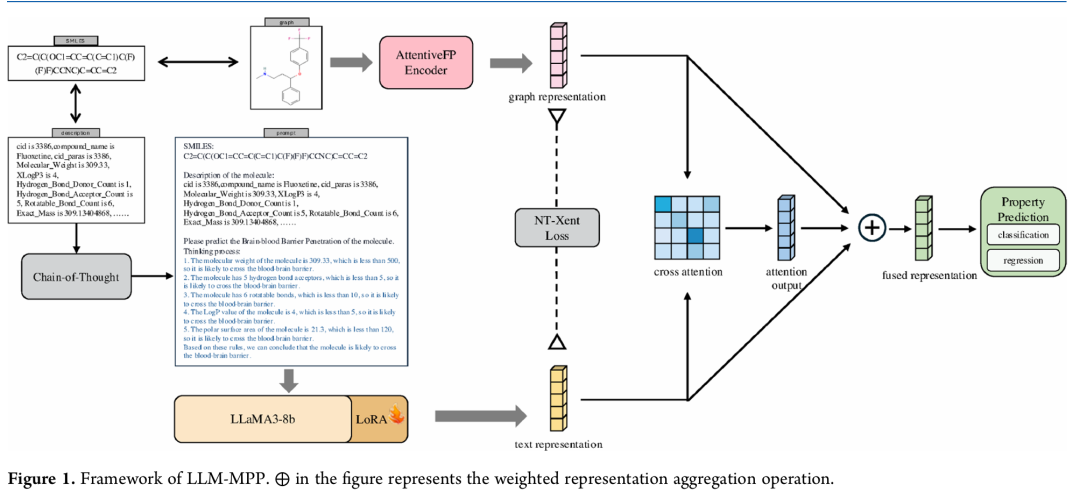
2. Molecular Property Prediction Based On Multi-Level Graph Self-Supervised Pretraining And Multi-Grain Finetuning
期刊: IEEE/ACM Transactions on Computational Biology and Bioinformatics 链接: https://doi.org/10.1109/TCBBIO.2025.3577899
简介: 本文针对分子性质预测中对多尺度化学结构难以同时建模的问题,提出了 MMGSF 框架。该框架首先在预训练阶段构建含原子、功能团(motif)及全图三类节点的分层分子图,并设计六个自监督任务:原子类型、键连通性与键类型生成,motif-atom 对比学习,以及原子数目和键数目预测,从而让 GNN 同时捕获局部到全局的化学语义信息。随后,在微调阶段引入 Multi-Grain Finetuning (MGF),分别利用均值池化、注意力池化和 MLP 得到原子粒、motif 粒及图粒表示,并通过可学习查询向量驱动的跨粒度注意力(mol-adapter)自适应融合,充分保留互补特征并抑制冗余。整体方法无需额外参数即可在训练过程中动态调节不同粒度信息的权重,从而构建更具判别力的分子表示。
实验中,作者使用 ZINC15 数据库 25 万条未标注分子进行自监督预训练,在 MoleculeNet 六个分类数据集(BACE、BBBP、Tox21、ToxCast、SIDER、ClinTox)以及 ESOL、FreeSolv 等回归数据集上评估。与 GraphCL、MolCLR、HiMol 等现有方法相比,MMGSF 在分类任务上取得 0.7407 的平均 ROC-AUC,优于最佳基线 0.7217;在 ESOL 等回归任务上亦将 RMSE 从 0.9419 降至 0.8745,显示了显著优势。消融研究进一步证明,去除任何一级自监督任务或粒度表示都会明显降低性能,验证了多层预训练与多粒度融合的互补性。总体而言,MMGSF 提供了一种兼顾结构完整性与语义表达的通用范式,为药物发现等场景的高效分子表征学习提供了新的思路。
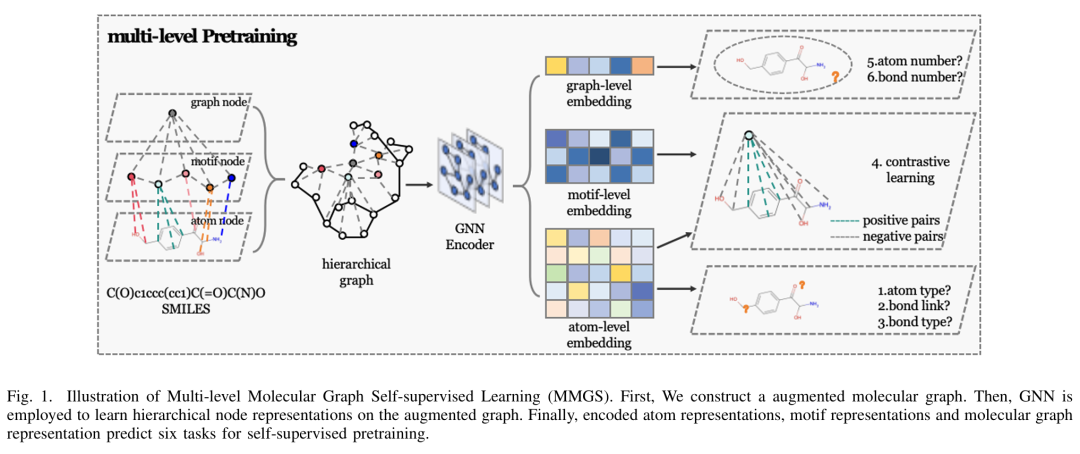
3. Contrastive Learning-Based Drug Screening Model For GluN1/GluN3A Inhibitors
期刊: Acta Pharmacologica Sinica 链接: https://doi.org/10.1038/s41401-025-01580-0
简介: 论文提出了 CLG-DTA,一种融合图对比学习与自然语言监督的新型药物-靶点亲和力预测框架,用于加速 GluN1/GluN3A 受体小分子筛选。模型包含药物图、蛋白质序列、亲和力文本三种模态的六个编码器,并将实验亲和力数值转写成英文词串,再借助共识数值知识图谱对连续文本嵌入施加顺序约束,从而获得判别性更强的分布式表示。在 Davis 与 KIBA 两个基准数据集上,作者设置四种冷启动情景 (S2–S4) 验证泛化能力;CLG-DTA 在多数场景的 PCC、MSE 等指标上优于 SVM、RF、MSGNN-DTA 和 HGRL-DTA 等方法,例如在 KIBA 的 “新药-新靶” 场景中 PCC 提升至 0.280,相比次优基线提高约 140%。消融实验显示,对比学习和 0.01 精度的数值文本化是性能提升的关键。 作者进一步将模型迁移到 BindingDB 中 3726 条 GPCR 亲和力数据,CLG-DTA 取得 MSE 1.148、PCC 0.734,显著优于最强对比 MSGNN-DTA (MSE 1.640、PCC 0.537)。在实际筛选中,模型对 1800 万化合物虚拟筛选后选出 12 个候选体,细胞水平验证发现 5 个活性化合物 (命中率 41.7%),其中 Boeravinone E 的 IC₅₀ 为 3.40 ± 0.91 μM,经膜片钳实验和分子对接确认其可能结合于 GluN1-GluN3A NTD 与 LBD 交界处。 总体而言,CLG-DTA 通过将数值亲和力语言化并与图结构特征对齐,实现了对陌生化学-生物空间的稳健外推,并在实验层面发现了新的 GluN1/GluN3A 抑制剂,为针对神经疾病的 NMDA 受体药物开发提供了有效工具。
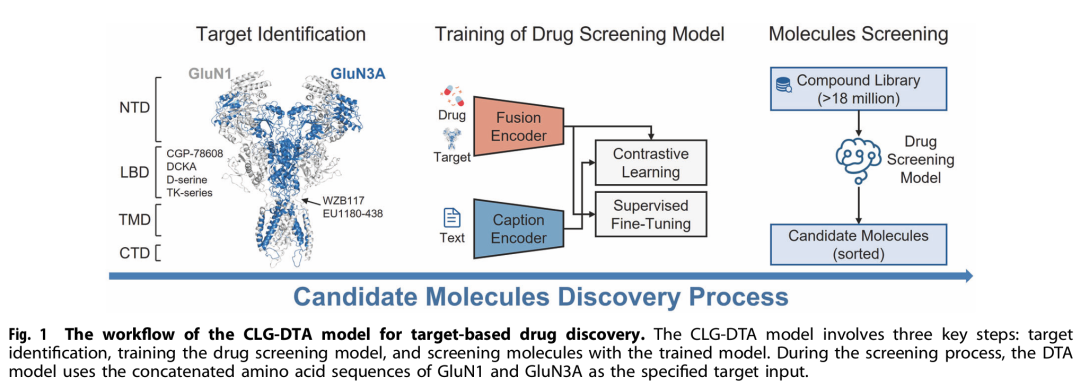
4. Advancing Ligand Binding Affinity Prediction With Cartesian Tensor-Based Deep Learning
期刊: bioRxiv 链接: https://doi.org/10.1101/2025.06.04.657800
简介: 论文提出了 PBCNet2.0,一种基于笛卡尔张量的等变图神经网络式成对比较架构,用于预测蛋白-配体相对结合亲和力。作者将训练集规模从前代的 60 万扩展到 860 万 BindingDB 复合物对,并在消息传递层引入 rank-2 笛卡尔张量,使模型能在不依赖预设势函数的情况下直接学习距离与角度信息,从而更精细地捕获蛋白-配体相互作用。在 16 个化学系列组成的 FEP 公开基准上,PBCNet2.0 的零样本 Spearman 相关系数达 0.67,逼近工业金标准 Schrödinger FEP+ 的 0.70,并显著超越 PIGNet2、RTMScore 等 10 种 AI 基线;少量 SAR 数据微调后,其排名精度进一步反超 FEP+,在 8 个真实项目回顾测试中将活性优化效率提升约 7 倍,同时节省近 40 % 资源。
为验证模型是否真正理解相互作用而非记忆配体特征,作者构建了 SAR-Diff、F-Opt 及 Mutation 三个难度更高的外部集合。PBCNet2.0 在所有 8 对 SAR-Diff 系列均保持有效预测,在针对含氟正交多极相互作用的 F-Opt 中获得最低 MAE 0.33 且 100 % 方向一致,并在 8 个临床相关靶点的突变集上取得平均 ρ = 0.53,显现出对耐药突变的外推能力。两项前瞻实验进一步印证了模型的实用性:对 ENPP1 抑制剂仅以 F→H 取代预测亲和力下降 40 倍,与 SPR 实测 ΔpK_D = 1.46 高度吻合;在 ALDH1B1 体系中准确指出手性异构导致的 10 倍活性差异,并通过计算扫描成功命中 5/6 关键耐药位点。作者亦讨论了静态构象与负样本稀缺的局限,并提出将大规模交互知识迁移到其他下游任务的展望。整体来看,PBCNet2.0 在保持高通量的同时将深度学习亲和力预测性能首次推至 FEP 级别,为探针发现和先导优化提供了高效且可解释的新工具。
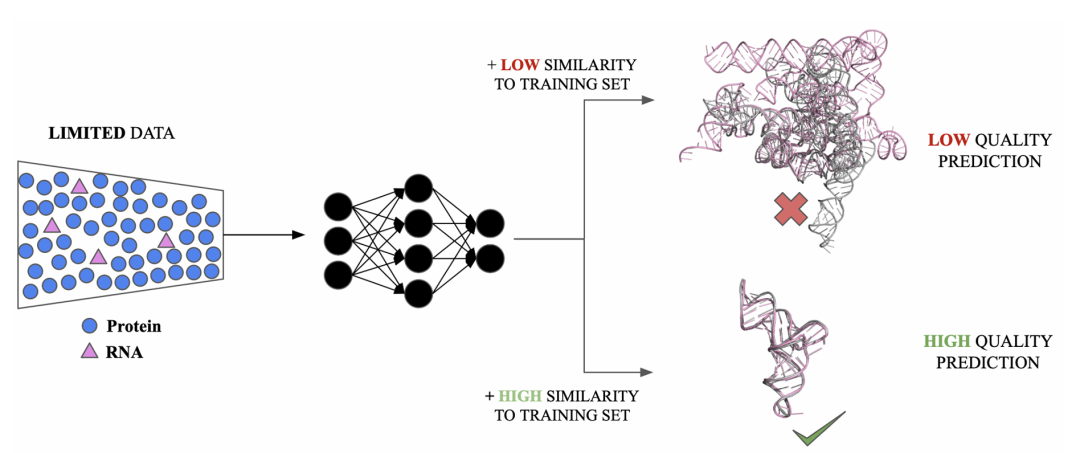
5. Limits Of Deep-Learning-Based RNA Prediction Methods
期刊: bioRxiv 链接: https://doi.org/10.1101/2025.04.30.651414 代码: https://github.com/iammarcol/RNA-Benchmark
简介: 作者针对 RNA 三维结构预测快速发展但普遍缺乏系统评测的现状,汇集了 AlphaFold3、Boltz-1、Chai-1、HelixFold3、NuFold、RhoFold+、RoseTTAFoldNA 和 trRosettaRNA 等九种最新深度学习模型,并在统一硬件与 MSA 设置下对其进行公平比较。研究者首先从 2021 年 9 月 30 日之后发布的 PDB 条目及 CASP16 目标中筛选出 50 条单链 RNA 与 42 个 RNA-RNA / RNA-蛋白复合物,过滤掉与 AlphaFold 训练集高度同源的序列与结构,以确保测试样本的独立性。评测使用重新校正适用于较短核酸链的 TM-score(d₀ 固定为 5 Å)评价单链模型质量,并结合 DockQ 评分量化复合体整体与界面准确度,同时检验各方法给出的 pTM/ipTM 置信度与真实精度的相关性。
结果表明,单链任务中 Boltz-1 取得平均 TM-score 0.512、成功率 53%,略优于 AlphaFold3 的 0.483 / 42%;复合体预测则以 AlphaFold3 表现最佳(TM-score 0.642、DockQ 0.395),Boltz-1 次之。进一步分析显示,模型性能与目标在训练集中的最大 TM-score 呈显著正相关,说明现有算法主要靠识别已见过的螺旋或三叶草等常见模体,而对新折叠的泛化能力有限;约三分之一的样本可获得高精度模型,若结构与训练数据相似度低于 0.25,平均 TM-score 降至 0.1 以下。此外,单链 RNA 的 pTM 置信度与实际 TM-score 相关性弱,ipTM 对识别复合体正确接口更具参考价值。作者指出,当前深度学习模型在 RNA 结构预测上仍受限于训练数据稀缺与准确度评估失真,未来需扩大多样化实验结构、改进模型不确定性量化,才能真正突破复杂或未知 RNA 构象预测的瓶颈。
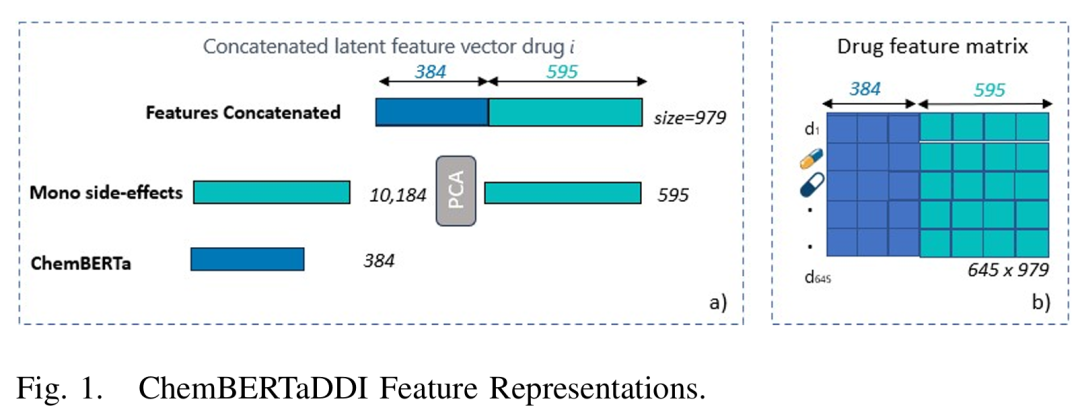
6. ChemBERTaDDI: Transformer Driven Molecular Structures And Clinical Data For Predicting Drug-Drug Interactions
期刊: bioRxiv 链接: https://doi.org/10.1101/2025.01.22.634309 代码: https://github.com/anastasiyagromova/ChemBERTaDDI
简介: 论文提出 ChemBERTaDDI 框架,通过将大型自监督语言模型 ChemBERTa-77M-MLM 生成的 384 维 SMILES 表征与 10184 维单药不良反应(mono side effects)经 PCA 降维后的 595 维临床特征拼接,构建 979 维多模态药物向量。对于每一对药物,作者采用元素级求和得到组合表示,再输入三层全连接网络完成 964 类多重不良反应的二分类预测。该方法在训练阶段仅微调预测器,保留 ChemBERTa 权重冻结,因而在捕获分子结构语义的同时避免额外计算开销。
实验以整合自 TWOSIDES、SIDER、OFFSIDES 和 PubChem 的 DB1 数据集为基准,覆盖 645 种药物、63473 对相互作用和 964 种多药并用不良反应。五折交叉验证显示,使用化学结构+单药不良反应特征的 ChemBERTaDDI 在 AUROC、AUPR 和 F1 分别达到 0.965、0.954 和 0.937,整体优于 Decagon、DeepDDI、MDF-SA-DDI、DPSP 与 NNPS 等五种主流方法;若仅保留化学结构信息则性能显著下降,证明临床特征对提升准确率至关重要。此外,模型在“恶性高血压”“子宫颈癌”等高危反应上的 AUPR 提升尤为明显,并保持对新药化合物的良好泛化能力。研究最后讨论了特征求和可能导致的信息损失及未来融入基因或蛋白组学数据的方向,为可扩展的 DDI 预测提供了新思路。
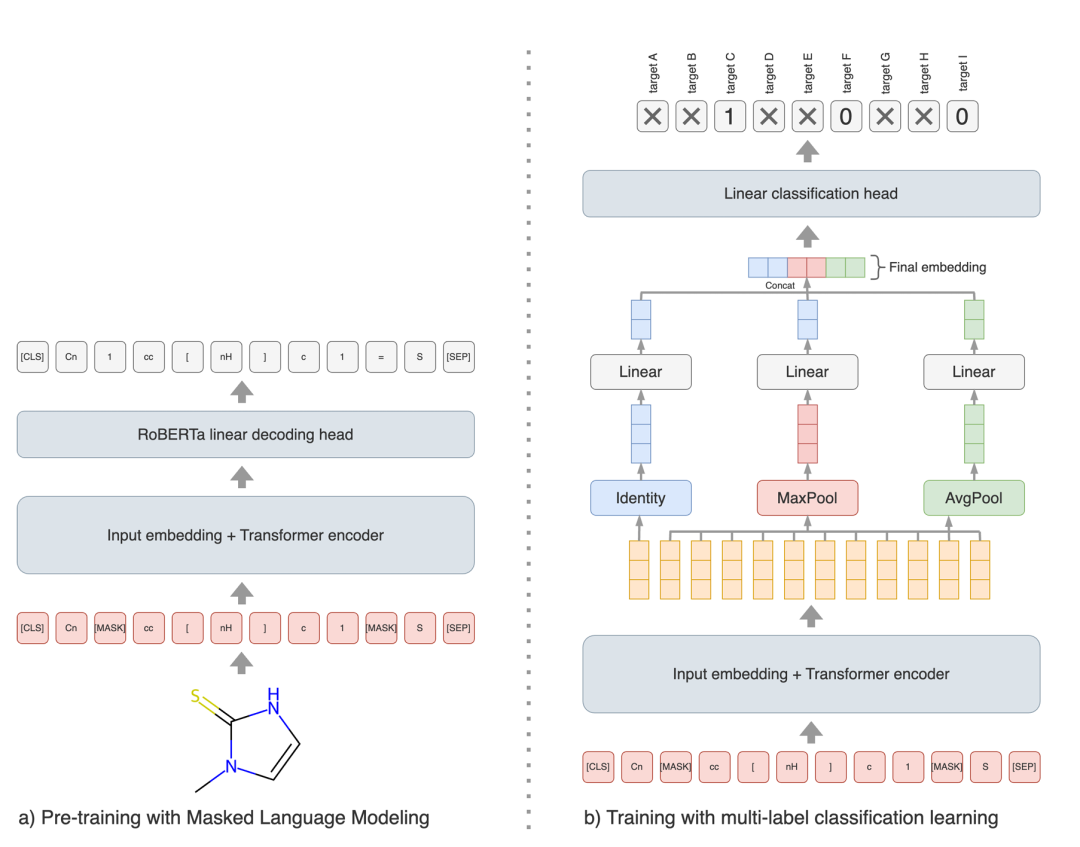
7. Bioptic B1: A Target-Agnostic Potency-Based Small Molecules Search Engine
期刊: arXiv 链接: https://arxiv.org/abs/2406.14572v4
简介: 作者提出 BIOPTIC B1,一种面向药物虚拟筛选的全局性、小分子活性检索模型。核心思想是在不依赖蛋白结构信息的情况下,仅用分子 SMILES 就学习“效力语义”嵌入,从而跨靶点发现结构完全不同但活性相近的候选化合物。模型首先在 1.6 亿条 PubChem 与 Enamine REAL Space 分子上进行 RoBERTa-式掩码语言模型预训练,随后利用 BindingDB 中约 6 700 个靶点的二元活性数据进行多标签微调,并通过多池化与 60 维归一化向量输出分子表示。嵌入检索采用 SIMD 加速的余弦相似度矩阵乘法,配合 27 台低配 CPU 节点和 270 块 SSD 构成的分布式索引,可在保持 100 % 召回率的同时,对 400 亿个分子库实现秒级查询。
在评估方面,作者针对七个不同蛋白靶点(如 ACHE、EGFR、JAK2 等)构建了 Hi-split 基准:保证测试分子与训练集的 ECFP4 相似度 < 0.4,以检验模型的脚手架跳跃能力。与 Chemprop、梯度提升树 (GB) 及传统指纹检索相比,BIOPTIC 在大部分靶点上取得更高或相当的 ROC-AUC、Average Precision 和 Precision@100;例如在 AA2AR,BIOPTIC 最优 AUC 为 0.744,而 Chemprop 为 0.685。检索速度测试显示:一次性为 40 B Enamine REAL Space 生成嵌入需约 5.6 h(8×A100 集群),之后单次查询在 SSD 模式下仅耗 2 min 15 s,而将嵌入置于内存可缩短至 44 s;整体系统可在纯 CPU 环境下于约 40 s 内返回 40 B 库的前 k 结果。
综上,BIOPTIC B1 将搜索引擎与推荐系统的工程最佳实践引入虚拟筛选,通过轻量级嵌入、硬件友好的检索算法和严格的“化学新颖性”评测框架,实现了对超大规模分子空间的实时、靶点无关活性发现,为大规模先导化合物挖掘提供了高效且成本可控的新途径。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-06-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号