Nat. Comput. Sci. | 基于大型扰动模型的计算生物学发现

Nat. Comput. Sci. | 基于大型扰动模型的计算生物学发现

DrugAI
发布于 2026-01-06 13:16:55
发布于 2026-01-06 13:16:55
DRUGONE
扰动实验通过测量扰动前后系统的变化,揭示生物系统的因果机制。这类数据包含了丰富的信息,可用于理解分子机制、基因与疾病的关系、以及药物开发。然而,不同实验的扰动类型、读出方式和生物背景差异极大,使得跨实验整合极具挑战。
研究人员提出了大型扰动模型(Large Perturbation Model, LPM),一种深度学习框架,用于整合多模态扰动实验。LPM将扰动(P)、读出(R)和实验上下文(C)表示为相互独立但可交互的维度。该模型在多个生物学任务中表现出色,包括:预测未见实验的扰动转录组、识别化学与遗传扰动的共享分子机制、推断基因–基因作用网络。LPM在统一的潜在空间中学习到生物学意义明确的联合表征,从而在计算机中实现生物关系的系统研究,并显著加速了大规模扰动实验的知识发现。

扰动实验是揭示生物系统因果机制的核心方法。通过在细胞或体内模型中引入特定基因或化合物扰动,研究人员可以比较扰动前后的读出差异,如转录本数量变化,从而解析分子机制、基因与疾病表型的因果关系。
随着高通量实验的普及,大规模扰动数据涵盖数千种扰动类型、不同读出方式(如转录组、表型、存活率)以及多样的生物背景(从单细胞到组织层级)。然而,这些数据在实验设计、测量尺度和生物模型上差异显著,导致跨实验整合困难。核心挑战在于:如何区分实验背景差异与扰动本身造成的效应。
传统计算方法多聚焦于预测未见扰动的效果,但其适用性受限于特定数据类型(如单细胞转录组)。近年来的基础模型(如Geneformer、scGPT)虽可通过Transformer编码器提取上下文信息,但受噪声影响大、难以兼容多模态数据。
因此,研究人员提出LPM,将扰动、读出与上下文分离建模,通过无编码器的解码结构实现多模态数据的统一整合与推理,支持多任务生物学发现。
方法概述
LPM的核心思想是:将每个扰动实验表示为三元组 (P, R, C),其中
- P 表示扰动类型(如基因敲降、药物作用);
- R 表示观测读出(如特定基因表达量或细胞存活率);
- C 表示实验上下文(如细胞系、实验条件)。
模型通过学习P、R、C三维嵌入空间的结构,预测任意组合下的实验结果。其架构为解码器结构(decoder-only),无需显式编码输入数据,具备以下特性:
- 异质性整合:支持不同实验类型、不同数据模态的融合学习。
- 上下文可分离表征:有效区分扰动效应与实验背景效应。
- 高扩展性:能在不同扰动组合下进行未见实验的预测。
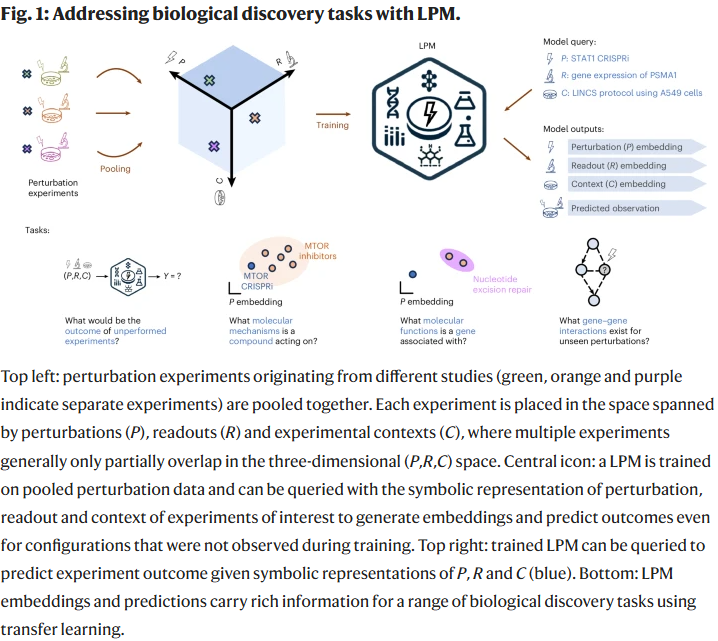
结果
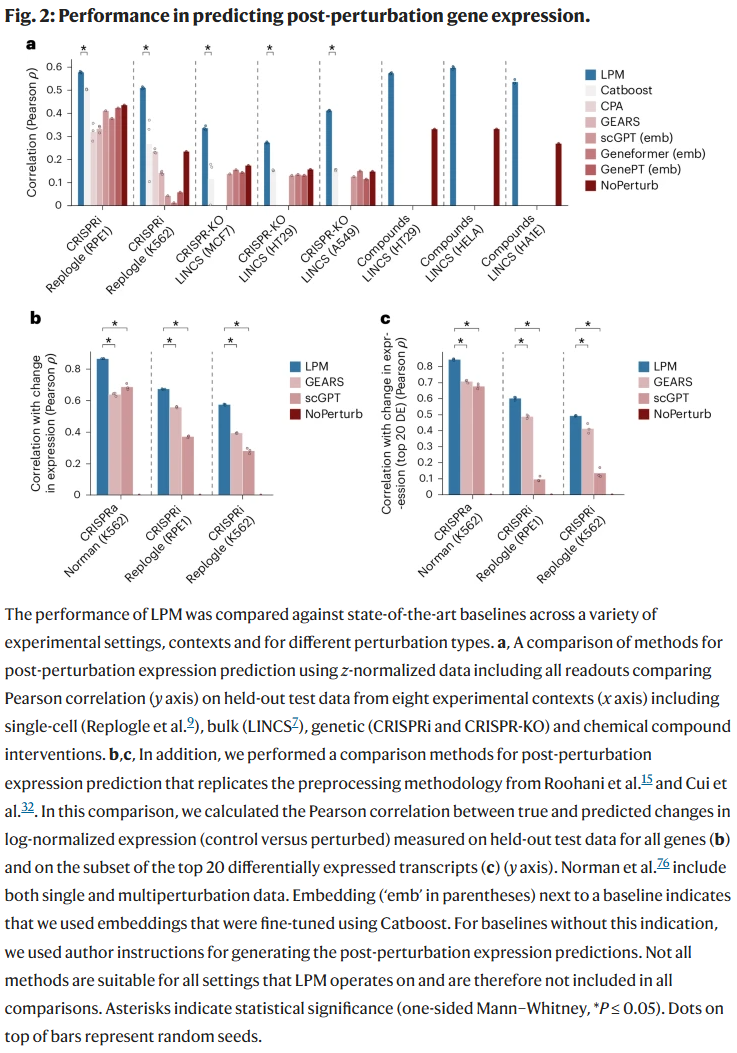
预测未见扰动实验的结果
研究人员在多组转录组扰动实验中比较了LPM与现有模型(包括CPA、GEARS、Geneformer、scGPT等)的表现。结果显示,无论是单细胞、bulk数据还是化学、遗传扰动,LPM在预测扰动后基因表达变化方面均显著优于其他模型。
在不同预处理方式和实验背景下,LPM的预测一致性和鲁棒性均更高,表现出跨模态泛化能力。
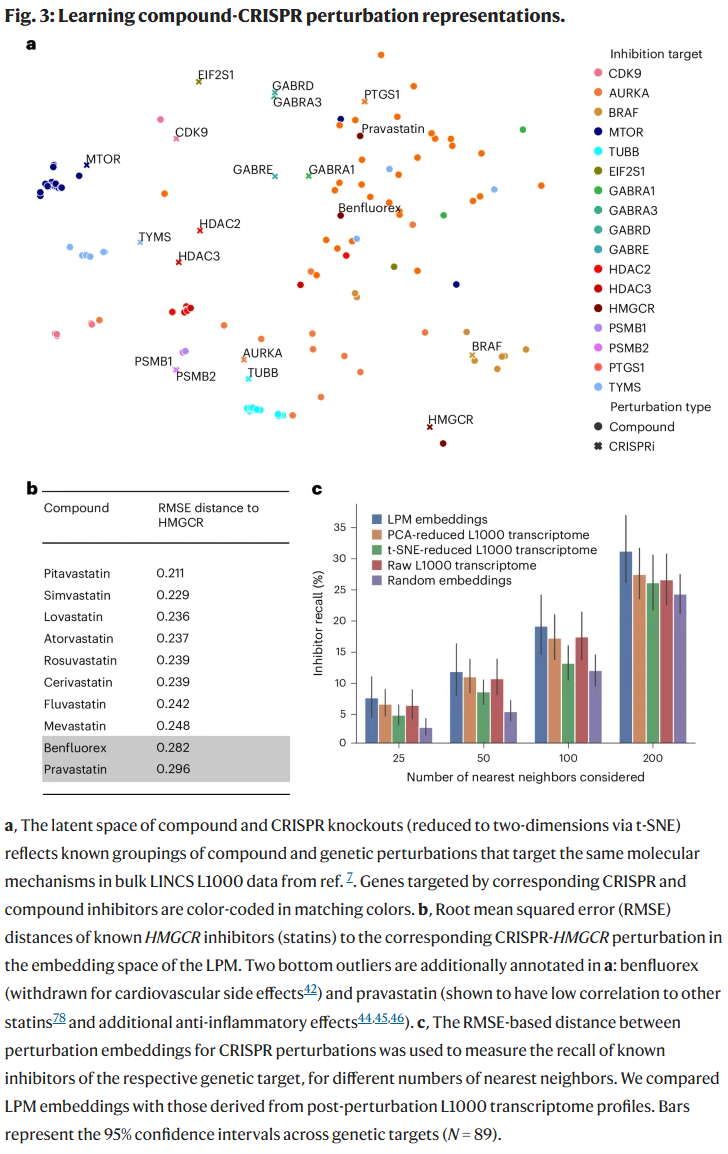
构建化合物–基因共享扰动空间
研究人员将LPM应用于LINCS数据库中的大规模化学与遗传扰动实验,统一学习两者的潜在空间。结果显示,药物抑制剂与其对应的基因CRISPR干扰常被聚类在相邻位置,例如MTOR抑制剂与MTOR基因敲降的扰动聚集在一起。
此外,LPM还揭示了药物的潜在作用机制,如pravastatin在模型空间中接近抗炎药PTGS1抑制剂,提示其可能具备抗炎效应。
与传统基于L1000转录组嵌入相比,LPM的潜在空间在识别药物靶点及同靶基因间关联上具有更高召回率。
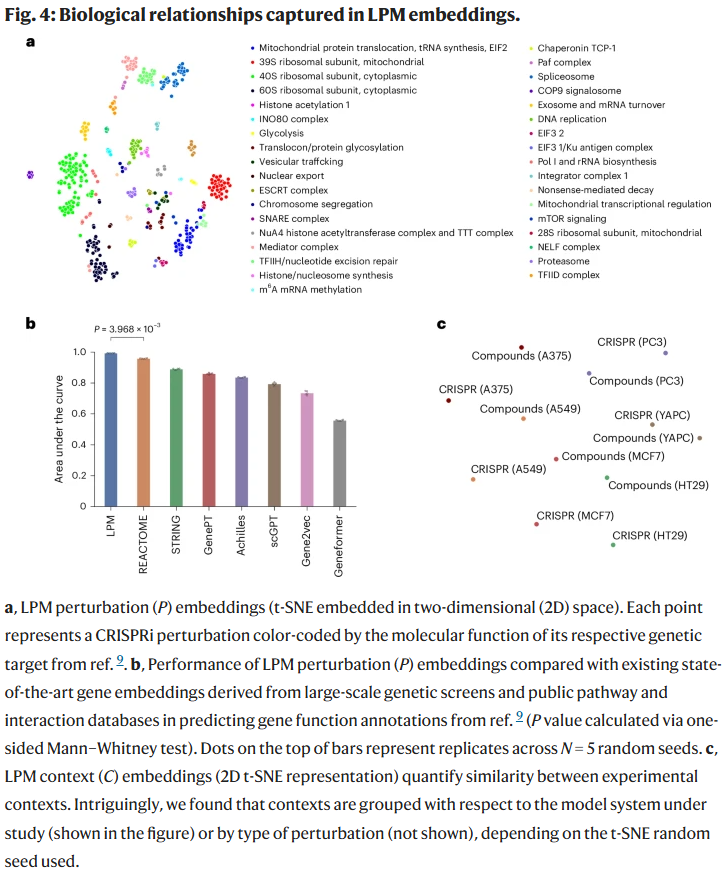
捕获已知的生物学关系
研究人员进一步验证了LPM学习到的扰动嵌入是否反映真实分子功能。结果表明,LPM自动将扰动按分子功能聚类,并在预测基因功能方面显著优于STRING、Reactome、Gene2Vec等传统数据库嵌入。
此外,通过t-SNE可视化,LPM学习到的上下文嵌入能自动区分实验系统(如不同细胞系),表明模型在潜在空间中捕获了生物语义信息。
在ADPKD中的计算药物发现
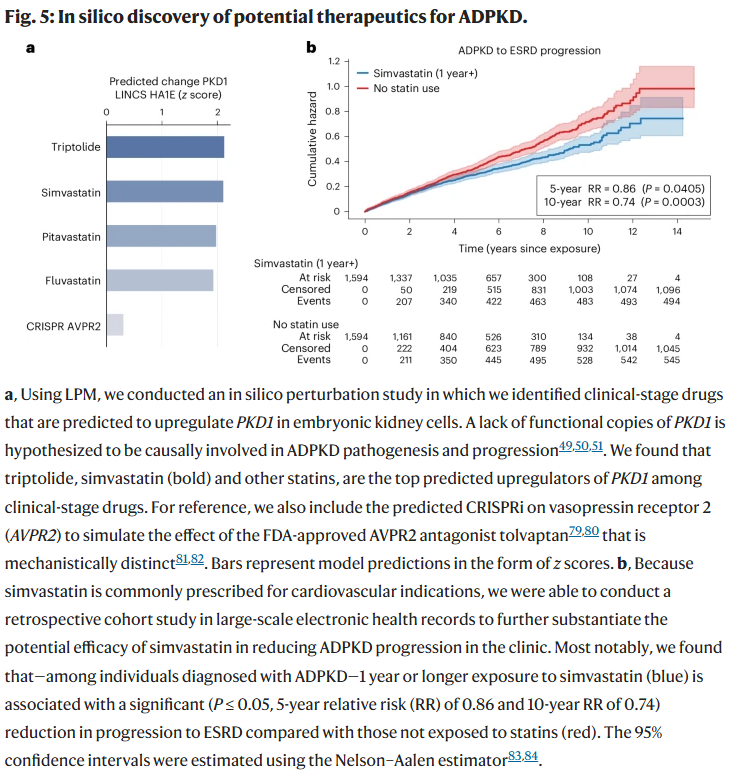
为展示LPM在药物再定位中的潜力,研究人员针对**常染色体显性多囊肾病(ADPKD)进行了计算筛选。
LPM在体外数据基础上预测能上调PKD1表达的临床药物,结果发现三七皂苷(triptolide)与辛伐他汀(simvastatin)**位列前列。随后利用真实世界健康记录进行回顾性验证,结果显示长期服用辛伐他汀的患者,其肾衰进展风险显著下降。这一研究展示了LPM在发现候选治疗药物方面的强大潜力。
推动基因–基因因果关系的推断
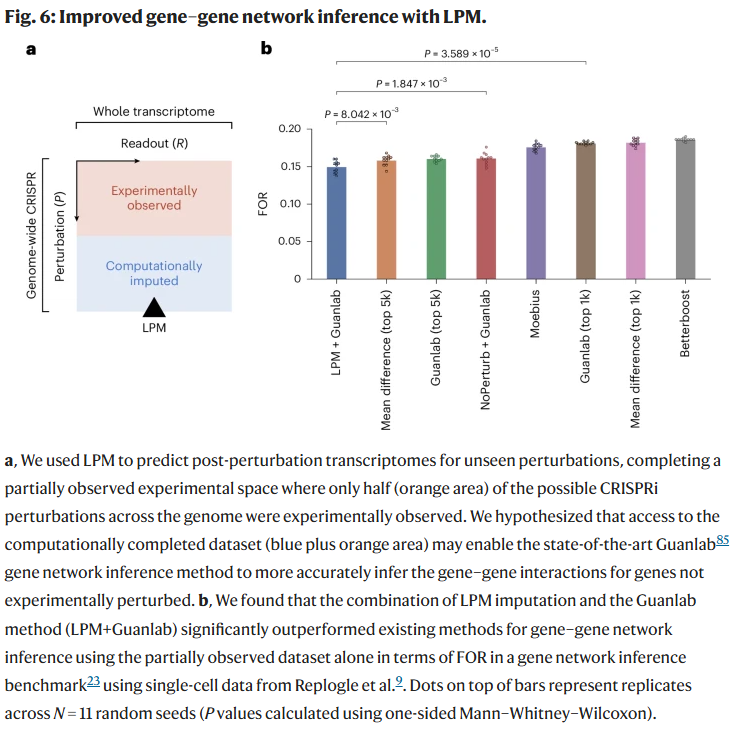
为了评估LPM预测结果在捕捉基因间机制性互作方面的能力,研究人员将LPM用于基因–基因相互作用网络的因果推断(causal inference)任务中。通常,这类网络是基于有限扰动实验构建的,因为实验条件下只对部分基因进行扰动。研究人员提出使用LPM在计算机中生成未观测的CRISPRi扰动数据,并将这些虚拟扰动结果与真实实验数据结合,用于增强因果网络的推断。
具体而言,研究人员采用在CausalBench挑战中表现最优的因果推断算法,对包含LPM预测数据的增强数据集进行网络推断。
结果显示,在原始实验数据中加入由LPM生成的推断性扰动结果后,推断得到的基因–基因网络在**假阴性率(False Omission Rate, FOR)**等指标上显著改善。这一结果表明,LPM不仅能补全未观测扰动实验,还能提升下游因果网络推断的准确性,说明其学习到的扰动–响应模式在统计上具有因果一致性。
讨论
LPM证明了整合异质扰动实验数据可在计算机中实现高精度生物学发现。它不仅能预测未观测实验的结果,还能揭示基因、药物与表型间的潜在机制。
主要优势包括:
- 统一性:可同时处理不同类型的扰动与读出;
- 可解释性:潜在嵌入空间反映真实生物学关系;
- 扩展性:预测性能随数据规模提升而显著增强。
然而,该框架仍有局限:目前主要基于标准化公共数据集,尚需在原代细胞、组织样本中验证;模型无法外推至训练词汇外的新符号(如全新基因或细胞类型);批次效应与元数据缺失仍可能影响性能。此外,ADPKD案例为回顾性研究,需进一步前瞻验证。
未来,通过与更多临床扰动数据整合,LPM有望成为推动个性化医学与分子机制发现的重要工具。
整理 | DrugOne团队
参考资料
Miladinovic, D., Höppe, T., Chevalley, M. et al. In silico biological discovery with large perturbation models. Nat Comput Sci (2025).
https://doi.org/10.1038/s43588-025-00870-1
内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-10-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号