文献解读-基于多种组装与 binning 工具的二代与三代宏病毒组比较评测:解析肠道病毒基因组功能特征

文献解读-基于多种组装与 binning 工具的二代与三代宏病毒组比较评测:解析肠道病毒基因组功能特征

用户1075469
发布于 2025-08-15 11:46:36
发布于 2025-08-15 11:46:36
近日,复旦大学与中国科学院微生物研究所等团队在《Microbiome》发表了一项重要研究,利用 95 份平行测序的人类粪便富集病毒样颗粒数据,系统评估了短读长测序、长读长测序及混合测序多种组装策略下多种病毒 binning 工具的效果与适用性。研究比较了 12 种组装软件在不同数据类型下的组装性能,发现 MEGAHIT、metaFlye 和 hybridSPAdes 在各自数据类别中表现最佳,研究还得对 4 种主流病毒 binner(MetaBAT2、CONCOCT、AVAMB 和 vRhyme) 在 HQ(高质量)与 NC(近完整)病毒基因组回收上的表现进行了全面比较。
结果显示,不同工具在不同数据类型与组装策略下的性能差异明显:有的更适合长读长数据,可大幅提升基因组连续性与完整性;有的在短读长数据中更擅长捕获低丰度病毒;还有的在混合组装结果中表现均衡,兼顾完整性与覆盖面。该研究是首次在大规模真实肠道病毒数据上系统评估多种病毒 binning 工具,明确了工具间的互补性与选择原则,为未来病毒基因组构建和分析流程优化提供了重要参考。
摘要
背景
宏基因组组装获得的病毒基因组(metagenome-assembled viral genomes, MAVGs)极大推动了人类肠道病毒组的发现与特征解析。然而,目前仍缺乏对不同组装工具在病毒基因组识别能力方面的系统性比较评估,尤其是针对二代测序(next-generation sequencing, NGS)与三代测序(third-generation sequencing, TGS)数据在解析效果上的跨平台评估。
结果
本研究评估了二代测序(next-generation sequencing, NGS)、三代测序(third-generation sequencing, TGS)及混合组装策略在病毒基因组发现中的性能。所用数据来自 95 份经病毒样颗粒(viral-like particle, VLP)富集的人类粪便样品,这些样品分别在 Illumina 平台和 PacBio 平台进行测序。评估结果显示,MEGAHIT、metaFlye 和 hybridSPAdes 分别是 NGS、TGS 与混合数据集的最优组装工具(assembler)。值得注意的是,这些组装工具在构建病毒基因组具有显著差异,体现出较高的互补性(complementarity)。与单一的组装工具相比,对不同组装工具结果进行整合后,非冗余高质量病毒基因组(high-quality viral genomes)的数量增加了 4.83 ~ 21.7 倍。在比较不同测序类型时,NGS 与 TGS 数据恢复的病毒基因组序列重叠最少,表明测序数据类型对病毒基因组恢复结果具有显著影响。此外,本研究还评估了四种 binning 方法(MetaBAT2、CONCOCT、AVAMB 和 vRhyme)。结果发现,CONCOCT 更容易将不相关的 contig(连续序列片段)聚类到同一 bin 中,而 MetaBAT2、AVAMB 和 vRhyme 在 bin 的包容性(inclusiveness)与分类学一致性(taxonomic consistency)之间表现出较好的平衡。
结论
本研究结果强调了基于宏基因组数据开展病毒发现所面临的挑战,并凸显了现有工具的局限性。研究建议在条件允许的情况下,结合多种组装工具(assembler)与不同测序技术,以获得更全面的病毒基因组信息。同时,结果也表明亟需开发适用于肠道病毒组(gut virome)组装的专用工具。本研究为推动肠道宏基因组背景下的病毒基因组研究提供了重要参考与实践指导。
方法
肠道病毒组分析中选择的宏基因组组装工具与 binning 工具
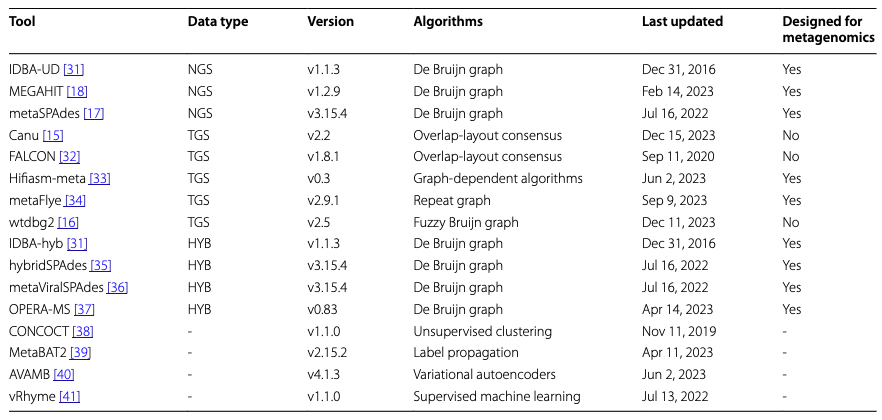
本研究为选择适用于肠道病毒组(gut virome)数据的最优组装工具(assembler)和 binning 工具,在分析中共纳入了 3 个短读长(short-read)组装工具、5 个长读长(long-read)组装工具、4 个混合组装工具(hybrid assembler)以及 4 个 binning 工具。所涉及到的工具和分析流程如Table 1 和Fig 1.
Table 1 Metagenomic assemblers and binners used in this study
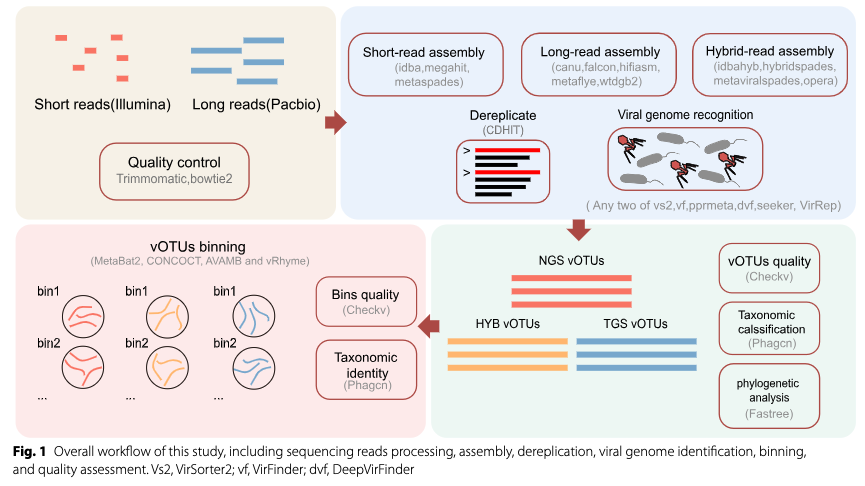
人类肠道病毒组样品进行Illumina 与 PacBio 平台测序
本研究使用了中国人类肠道病毒组(Chinese Human Gut Virome, CHGV)数据集中的测序数据,该数据集包含来自 95 名中国健康居民的粪便样品,并分别采用短读长(short-read)和长读长(long-read)测序技术进行分析。
简而言之,粪便样品(每份约 500 g)采集自武汉和上海的匿名健康志愿者。病毒样颗粒(viral-like particles, VLPs)通过改良的病毒组富集流程分离:将 400–500 g 冷冻粪便(−80 °C 保存)加入 5 L SM 缓冲液(200 mM NaCl、10 mM MgSO₄、50 mM Tris–HCl,pH 7.5),在室温下使用自动搅拌器(A200plus, OuHor, 上海,中国)以 120 rpm 搅拌直至完全分散。混合液通过四层纱布(21 s × 32 s / 28 × 28)过滤,并在 4 °C 下依次以 5000 × g 和 8000 × g 分别离心 45 min。上清液经 100 kD 超滤膜(Sartorius, Vivaflow 200)浓缩至约 300 ml,加入 NaCl 至终浓度 0.5 M,4 °C 静置 1 h。随后加入 PEG 8000 至终浓度 10%(w/v),于 4 °C 过夜孵育,并在次日以 13,000 × g 离心 35 min(4 °C)沉淀噬菌体颗粒。核酸提取采用 HiPure HP DNA Maxi Kit(D6322, Magen, 广州,中国)按照说明书操作。所得双链 DNA(double-stranded DNA, dsDNA)分别在 Illumina HiSeq2000 平台(Novogen, 北京,中国)进行二代测序(next-generation sequencing, NGS),以及在 PacBio RS II 平台(Pacific Biosciences, Menlo Park, CA, USA)进行三代测序(third-generation sequencing, TGS)。
预处理VLP 测序数据
对于二代测序(next-generation sequencing, NGS;短读长)原始数据,采用 Trimmomatic(v0.39)去除测序接头(adaptor)并过滤低质量碱基,参数设置为 LEADING:3、TRAILING:3、SLIDINGWINDOW:15:30、MINLEN:50;对于三代测序(third-generation sequencing, TGS;长读长)数据,使用 pbccs(v4.0.0)在默认设置下进行错误校正(见图 1)。为检测修剪后的短读长数据或经 CCS(circular consensus sequencing)处理的长读长数据中可能来源于人类的 reads,使用 Bowtie2(v2.4.2)将修剪后的短读长数据与经 CCS(circular consensus sequencing)处理的长读长数据比对到人类参考基因组,以识别其中可能来源于宿主的 reads。
组装VLP 测序数据
将各个样本的 VLP 测序数据分别使用预先遴选的组装工具(assembler)进行组装,除特别说明外均采用默认参数。具体而言,NGS(短读长)数据使用 IDBA-UD、MEGAHIT 和 metaSPAdes 进行组装;TGS(长读长)数据使用 Canu、FALCON、Hifiasm-meta、metaFlye 和 wtdbg2 进行组装;短读长与长读长联合的混合组装(hybrid assembly)则使用 IDBA-hyb、hybridSPAdes、metaViralSPAdes 和 OPERA-MS(详见图 1)。
随后,利用 metaMIC(默认参数)对各组装工具生成的 contig(连续序列片段)进行误组装(mis-assembly)检测。对判定为误组装的 contig,按照 metaMIC 报告的误组装位点进行断点切分与校正,所得片段视作 contig 并纳入后续分析。
Contig 去冗余与病毒识别contig
对每个样本中各组装工具所得 contig,或多个工具在所有样本中所得 contig,使用 cd-hit(v4.6.8)按照 MIUViG标准进行去冗余处理(参数:-c 0.95,-aS 0.85)(见图 1)。随后,参考 Human Gut Virome Database(GVD)的分析流程并进行适当调整(见图 1)识别病毒 contig。首先,使用以下病毒识别软件:VirSorter2(v2.2.4)、DeepVirFinder(v1.0)、VirFinder(v1.1)、Seeker、PPR-Meta(v1.1)和 VirRep。参数设置如下:
- VirSorter 评分 ≥ 0.7
- DeepVirFinder:pvalue<0.05
- VirFinder 评分 > 0.6
- Seeker:默认参数
- PPR-Meta 噬菌体(phage)评分 > 0.7
- VirRep:默认参数
当某一 contig 在上述六项标准中至少满足两项,且序列长度大于 1.5 kb 时,将其判定为病毒 contig。最终,识别出的病毒 contig 按菌株水平(strain level)划分为病毒操作分类单元(viral operational taxonomic units, vOTUs)。
病毒contigs binning
在组装完成后,对各样本中已识别的病毒 vOTUs 进行多覆盖度(multi-coverage)binning。该方法在将某一样本的 contig 聚类为 bin 时,不仅考虑该样本自身的 reads 覆盖度,还同时参考这些 contig 在所有样本中的覆盖度信息。使用 CONCOCT、MetaBAT2、AVAMB 和 vRhyme,在默认参数下生成 bin(见图 1)。
注:multi-coverage binning--binning 策略的一种优化形式。常规 binning 通常只用单一样本的 contig 覆盖度信息,而 multi-coverage 会引入跨样本的覆盖度数据,这有助于更准确地将 contig 聚到正确的 bin 中,尤其是在不同样本中病毒丰度差异较大时。
vOTUs 与 bin 的质量评估
为评估 vOTUs 的质量,使用 CheckV(v1.0.1)(见图 1,参数:end_to_end),将 vOTUs 按完整度(completeness)划分为“complete”(完整,100%)、“high quality”(高质量,>90%)、“medium quality”(中等质量,50–90%)、“low quality”(低质量,0–50%)以及“not determined”(未确定)。在本研究中,将完整度 >90%,且未出现 “contig >1.5 × longer than expected genome length”(contig 长度超过预期基因组长度 1.5 倍)和 “high kmer_freq may indicate large duplication”(高 k-mer 频率可能提示大规模重复)警告信息的 vOTUs 定义为高质量 vOTUs(high-quality vOTUs,hq-vOTUs)。
目前尚无专门用于病毒 bin 的质量评估工具,且 CheckV 只能接受 contig 作为输入。因此,本研究借鉴了一种方法,即将 bin 内所有 contig 以 50 个连续的“N”字符连接为一条序列(CheckV 会将“N”视为缺口而非最短序列长度),再使用 CheckV 对其进行质量评估。
物种注释与系统发育分析
本研究使用 PhaGCN_newICTV 对所有高质量 vOTUs(hq-vOTUs)进行科水平(family-level)分类学注释(见图 1),并仅保留 PhaGCN_newICTV 评分为 1(评分范围 0–1)的结果作为最终注释,以确保分类结果的可靠性。在系统发育分析中,首先使用 Prokka(v1.14.6)对选定 vOTUs 的蛋白编码基因进行功能注释,并提取其中的terminase 大亚基(large terminase subunit, TerL)基因及其蛋白序列。随后,利用 MUSCLE(v3.8.1551)对这些蛋白序列进行多序列比对,并将比对结果输入 FastTree(v2.1.11),采用最大似然法(maximum-likelihood algorithm)构建系统发育树(见图 1)。最后,使用 iTOL(v6.8)与 Evolview v3 对系统发育树进行可视化和注释。
主要结果
基于短读、长读和混合测序数据,评估并鉴定 vOTU 检测的最佳组装工具
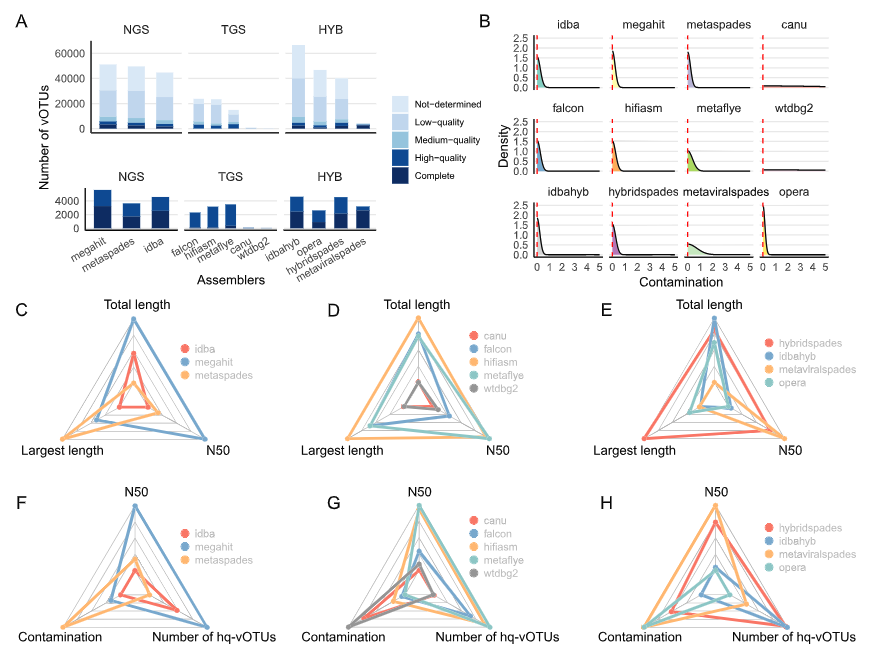
Fig.2|主要比较了不同组装工具在短读(NGS)、长读(TGS)和混合测序(HYB)条件下用于 vOTU(viral Operational Taxonomic Units)检测的效果,从数量、质量和多项统计指标进行综合评估。
A. 堆叠柱状图显示不同组装工具得到的 vOTU 数量,上方为全部 vOTU,下方为高质量 vOTU(hq-vOTU,完整度>90%、无大规模重复等)。不同颜色代表 CheckV 评估的质量等级(低质量、中质量、高质量、完整等)。 B. 密度图显示各组装工具得到的 vOTU 污染度分布(CheckV 结果),红色虚线为预期的污染阈值线。 C-E. 雷达图比较 NGS、TGS 和 HYB 三类组装工具在总组装长度、最大 vOTU 长度和 N50 等指标上的表现。 F-H. 与 C-E 类似,但比较的指标是 N50、0 污染 vOTU 比例,以及高质量 vOTU 数量。
结论:综合分析表明,MEGAHIT、metaFlye 和 hybridSPAdes 分别是 NGS、TGS 和混合组装类别中表现最优的工具,能够在保证更高质量的同时鉴定出数量更多、长度更长的 vOTU。
综合多种组装工具可在不同测序策略下获得更高质量的病毒基因组
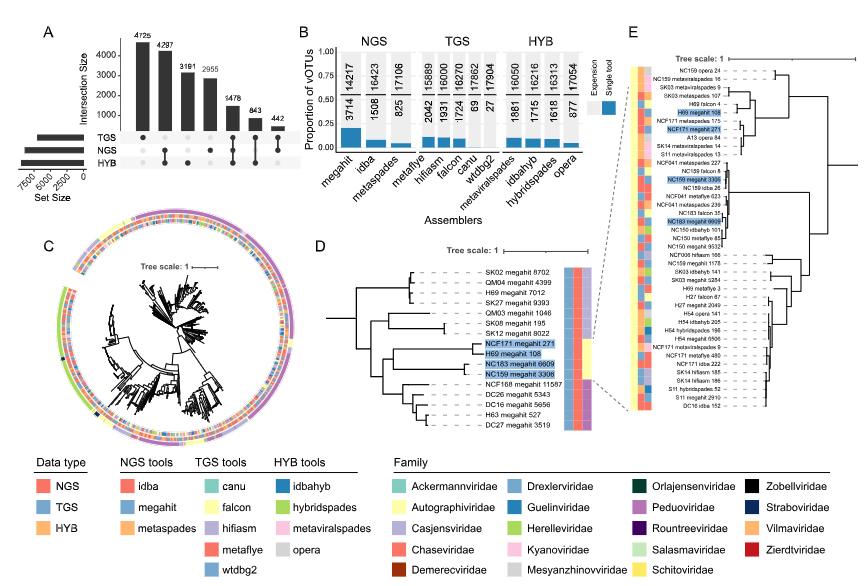
Fig.3|主要说明了不同组装工具在高质量 vOTU(hq-vOTU)恢复上的互补特性,以及这些 hq-vOTU 的系统发育关系与分类分布情况。
A. Upset 图展示了来自 NGS、TGS 和混合组装(HYB)三类工具的 hq-vOTU 数量及交集分布。结果表明,大部分 hq-vOTU 仅由某一类工具独立获得,不同测序策略与组装工具在产出上存在显著互补性。
B. 堆叠柱状图对比了各组装工具独立组装得到的 hq-vOTU 数量(深蓝色及下方数字)与通过其他工具结果扩展了 hq-vOTU 数量(浅灰色及上方数字),不同工具在 hq-vOTU 产出能力上差异明显。
C. 基于 1026 个编码大型末端酶基因的 hq-vOTU 构建系统发育树。叶节点标签旁的三层同心圆依次表示:组装工具类型、测序数据类型(NGS、TGS、HYB)以及科水平的分类注释。
D. 由 megahit(NGS 数据)特异组装的部分 hq-vOTU 构建的系统发育树,其中蓝色高亮的分支归属于 Autographiviridae 科。
E. 所有归属于 Autographiviridae 科的 hq-vOTU 系统发育树(去冗余合并集),蓝色背景标记的分支为 megahit 结果。左侧热图依次表示门水平注释(PhagCN_newICTV 工具)、数据类型以及组装工具。
结论:多组装工具策略可显著扩展肠道病毒组鉴定范围,并提高病毒基因组的覆盖度与多样性。
不同组装工具在不同分类层级(如科/目)鉴定 vOTU 的偏好性
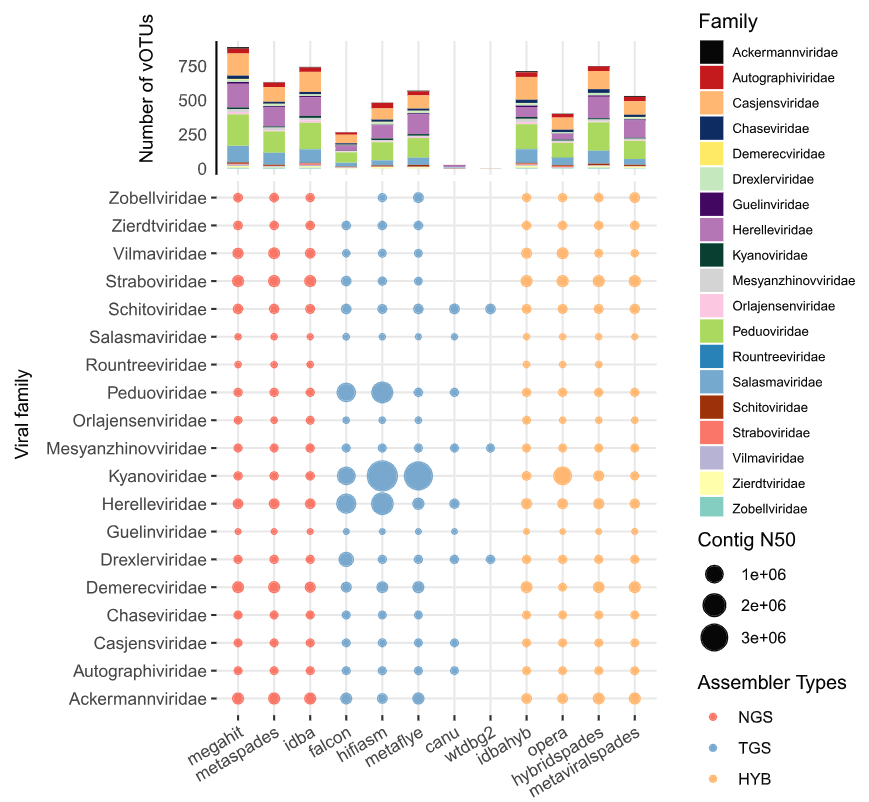
Fig.4|主要比较了不同组装工具在恢复和组装各病毒科 vOTU 时的分类表现与组装质量差异。
结论:不同组装工具在不同分类学水平鉴定病毒 contig 时存在偏好性,尤其是 TGS 组装工具表现出更明显的差异。
不同 binner 的 vOTU binning 性能与偏好比较
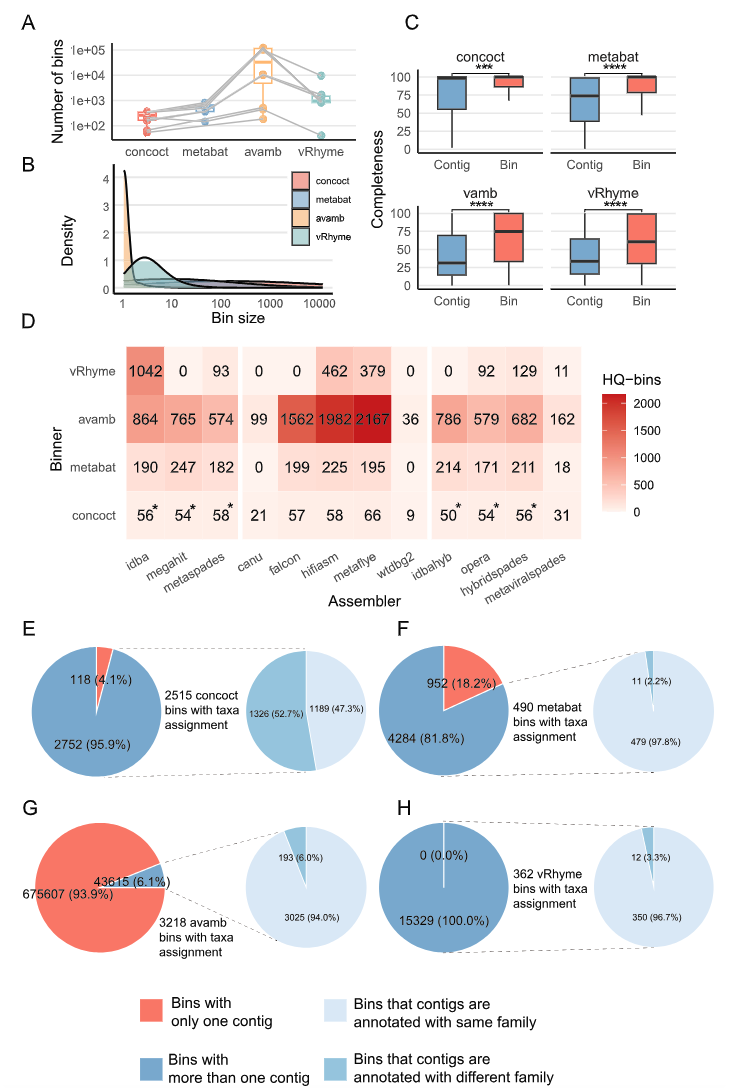
Fig.5|主要比较了四种 binning 工具(CONCOCT、MetaBAT、AVAMB、vRhyme)在不同组装工具生成的 vOTU 基础上进行病毒基因组 binning 的效果,从 bin 数量、大小分布、完整度提升、高质量 bin 数量以及分类一致性等方面进行了系统评估。
A. 箱线图显示四种 binning 工具在不同组装工具数据上的 bin 数量分布,AVAMB 和 MetaBAT 生成的 bin 数量相对较多。 B. 密度图展示了四种工具生成的 bin 大小分布,大多数 bin 的 vOTU 数量较少,但 AVAMB 在大 bin 生成上占比较高。 C. 箱线图比较了 binning 前(单个 contig)与 binning 后(bin)的完整度,四种工具均显著提升了完整度(Wilcoxon 检验,p<0.001 或 p<0.0001)。 D. 热图显示了高质量 bin(完整度>90%、无超长 contig 或高 k-mer 频率警告)的数量分布,AVAMB 和 MetaBAT 在多个组装工具数据上生成的高质量 bin 数量最多。 E-H. 饼图展示了各工具生成的 bin 内部 contig 在分类学上的一致性。
结论:比较了四种 binning 工具(CONCOCT、MetaBAT2、AVAMB、vRhyme)在 vOTU 数据上的表现,涉及 bin 数量、完整度提升、高质量 bin 产出 以及 分类注释一致性:
- 数量表现:AVAMB 在所有组装工具数据上生成的 bin 数量最多;CONCOCT 生成的 bin 内 contig 数量显著高于其他工具。
- 完整度提升:所有工具均能显著提高 bin 完整度,其中 AVAMB 提升幅度最大,其次是 vRhyme 和 MetaBAT2;CONCOCT 提升最小。
- 高质量 bin 产出:AVAMB 生成的高质量 bin 数量在四种工具中最多。
- 分类一致性:MetaBAT2、AVAMB 和 vRhyme 的 bin 内分类一致性高(>94%),而 CONCOCT 的一致性最低(约 47% 不一致),且更倾向于将不同科的 vOTU 聚在一起。
结论
根据95 份人类粪便富集病毒样颗粒(VLP)的配对长读与短读测序数据,开展了覆盖原始数据质控、组装、binning、病毒序列鉴定与分类注释的系统性分析。对 12 种组装工具与 4 种 binning 工具的评估显示,MEGAHIT、metaFlye 与 hybridSPAdes 在各自数据类型分组中表现优异。不同 binning 工具在多项指标上存在显著性能差异。此外,不同组装工具与数据类型生成的 vOTU 呈现高度互补性与差异性。上述结果强调,在病毒组数据分析中,应采用多工具联合并整合多种数据类型,以高效重建病毒基因组。
本研究构建的分析流程(包括数据集与性能评估体系)可方便地用于测试任何新工具,小编觉得这篇文章的亮点在于评估了不同binner工具对病毒binning效果。
数据集
本研究生成的原始测序数据已上传至 CNCB GSA 数据库,登录号为 PRJCA008836:
- GSA 链接:https://ngdc.cncb.ac.cn/gsa/browse/CRA006494
- BioProject 页面:https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA008836
- 由各组装工具生成的病毒 contig 的 FASTA 文件已上传至 Figshare:https://figshare.com/articles/dataset/Viral_contigs_of_Virome_Benchmark/25060193
参考文献
Wang H, Sun C, Li Y, Chen J. et el .Complementary insights into gut viral genomes: a comparative benchmark of short- and long-read metagenomes using diverse assemblers and binners. Microbiome. 2024 Dec 20;12(1):260. doi: 10.1186/s40168-024-01981-z. PMID: 39707560.
关于小编
小编就职于中国热带农业科学院,环境与植物保护研究所农业农村部热带地区低碳绿色农业重点实验室。目前实验室主要以研究方向是使用宏基因组学、宏病毒组等多组学研究微生物对土壤,大气等介质中元素循环的作用,环境中微生物功能基因的挖掘。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号