上下调基因分开注释就应该有生物学功能的重叠


昨天我们在推文:做生信压根就不怕造假或者出错 留下来了一个学徒作业, 就是读取作者给出来的表达量矩阵文件(GSE273142_Processed_data.tab.gz),完成合理的差异分析后,针对上下调基因进行独立的富集分析,然后看作者文章里面的EMT通路的变化情况哈!
之所以有这样的一个作业,其实是因为这里面又蕴涵着另外一个大家很容易弄错的知识点,就是如何理解生物学功能的上下调问题,文章里面的EMT通路的GSEA如下所示 :
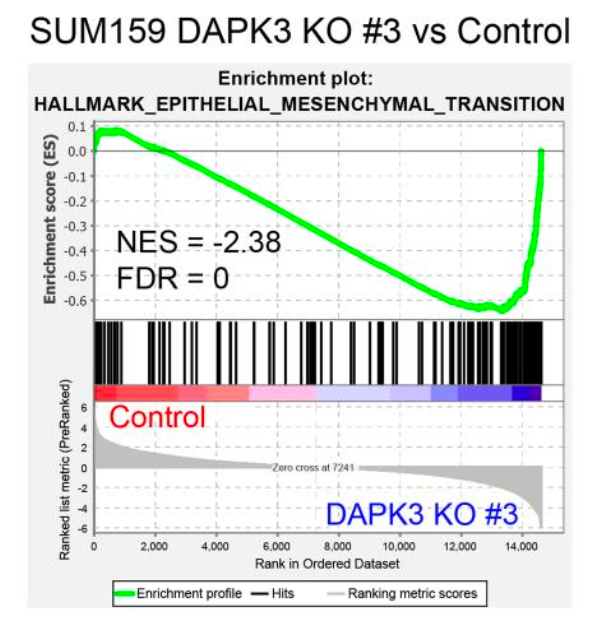
EMT通路的GSEA
我们可以让人工智能大模型帮我们介绍一下上面的图表:
这个图是一个基因集富集分析(Gene Set Enrichment Analysis, GSEA)的结果图,通常用于确定在两个不同条件下(如实验组与对照组)基因表达模式的差异是否显著。在这个例子中,比较的是SUM159细胞系中DAPK3基因敲除(KO #3)与对照组的差异。
图的解释:
- 标题:图的标题“SUM159 DAPK3 KO #3 vs Control”表明这是在SUM159细胞系中DAPK3基因敲除(KO)与对照组的比较。
- 富集图(Enrichment plot):显示了基因集在排序数据集中的分布。绿色曲线代表富集得分(Enrichment Score, ES),它反映了基因集在排序列表中的位置。
- NES(Normalized Enrichment Score):标准化富集得分,用于衡量基因集在两个条件下的表达差异。在这个图中,NES = -2.38,表示DAPK3 KO #3组在“HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION”基因集中的表达显著低于对照组。
- FDR(False Discovery Rate):错误发现率,FDR = 0 表示没有假阳性结果,即所有显著的富集都是真实的。
- 基因分布(Hits):黑色竖线表示基因集中的基因在排序列表中的位置。红色和蓝色条形表示基因在对照组和DAPK3 KO #3组中的表达水平。
- 排名指标(Ranked list metric):灰色曲线表示基因在排序列表中的排名指标,通常与基因表达的差异有关。
GSEA的原理:
GSEA是一种计算方法,用于确定在两个生物状态之间一个先验的基因集是否表现出显著的表达变化。它通过以下步骤进行:
- 基因排序:基于某种统计量度(如t-test, log2 fold change)对基因进行排序。
- 富集得分计算:计算基因集在排序列表中的运行得分,以评估基因集在两个状态下的表达差异。
- NES计算:通过标准化富集得分,以考虑基因集大小和基因排序列表的长度。
- 显著性评估:通过置换测试或类似方法评估NES的显著性。
如何解读GSEA结果:
- NES值:NES值越远离0,表示基因集在两个条件下的表达差异越显著。正值表示基因集在实验条件下上调,负值表示下调。
- FDR:FDR值越低,表示结果的可靠性越高。
- 排名指标曲线:曲线的形状和位置可以提供关于基因集在两个条件下表达模式的直观理解。
- 基因分布:查看基因集中的基因在排序列表中的分布,可以了解哪些基因对富集得分贡献最大。
通过这些信息,研究者可以识别在特定生物学条件下显著变化的基因集,进而推断可能的生物学机制或疾病过程。
EMT 背景知识
同样的,也是让人工智能大模型帮我们梳理这方面背景知识:
EMT,全称epithelial–mesenchymal transition,又称翻译为上皮间质转换,指的是上皮细胞在一些因素的作用下,失去极性及细胞间紧密连接和黏附连接, 获得了浸润性和游走迁移能力, 变成具备间质细胞形态和特性的细胞的改变。这种行为是可逆的。
EMT的概念最早是1982年由Green-berg和Hay提出。然而,长久以来,科学界对于EMT在肿瘤转移过程中的作用一直存在争议,主要是因为无法在体内观察EMT过程 。胚胎发育与癌症发展中的细胞可塑性变化有着惊人的相似性,而这种可塑性变化受到上皮间质转化epithelial-mesenchymal transition (EMT)过程的调节。胚胎发育时期,上皮状态和间充质状态的细胞能够自由转化。
- 上皮间质转化(EMT)使得细胞具备转移和浸润特性。
- 其反向过程,间质上皮转化mesenchymal-epithelialtransition (MET)赋予了细胞极性变化并失去移动能力。
EMT会促发癌细胞从病灶分离,转移到其它部位,而MET导致癌细胞停留,并在停留处引起新的肿瘤。
更多的EMT打分算法
上面的EMT通路的GSEA结果只能说看到DAPK3 KO #3组在“HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION”基因集中的表达显著低于对照组,并不能区分上皮细胞(epithelial cells)和间充质细胞(mesenchymal cells)之间的转换方向。这里我们推荐一个预印本文章,对3种EMT打分算法进行了测评,挺有意思的,标题是:《Comparative study of transcriptomics-based scoring metrics for the epithelial-hybrid-mesenchymal spectrum》,链接在 https://www.biorxiv.org/content/10.1101/2020.01.02.892604v1.full
分别是:
- 加权线性加和(76-gene EMT signature) 这个算法对CDH1 (E-cadherin)的权重很高,所以epithelial的EMT scores是比mesenchymal高。 参考文献:An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res.
- two-sample Kolmogorov-Smirnov test This score varies on a scale of −1 to 1, with the higher scores corresponding to more mesenchymal samples (Tan et al., 2014). 参考文献:Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol. Med.
- multinomial logistic regression 它的值集中于0到2之间, This method particularly focuses on characterizing a hybrid E/M phenotype using the expression levels of 23 genes – 3 predictors and 20 normalizers – identified through NCI-60 gene expression data. 会把样品判断为3种状态:epithelial, mesenchymal, or hybrid E/M categories
如果我们选择上下调基因分开注释
衔接前面的推文:做生信压根就不怕造假或者出错 里面的差异分析,然后需要首先根据一系列统计学指标,比如p值以及变化倍数来人为的规定好统计学显著的上下调基因列表,然后去进行超几何分布检验:
if(T){
nrDEG=DEG_deseq2[,c("log2FoldChange", "padj")]
#nrDEG=DEG_limma_voom[,c("logFC", "adj.P.Val" )]
head(nrDEG)
colnames(nrDEG)=c('logFC','P.Value')
# 凡是阈值,都是可以自定义
logFC_t <- 1
pvalue_t <- 0.1
gene_up= rownames( nrDEG[with(nrDEG,
logFC > logFC_t & P.Value < pvalue_t),])
gene_down=rownames( nrDEG[with(nrDEG,
logFC < -logFC_t & P.Value < pvalue_t),])
}
library(org.Hs.eg.db)
library(clusterProfiler)
# 一本书,很多数据库,很多可视化
library(ggplot2)
library(stringr)
## 2.3.1 KEGG----
{
kk.up <- enrichKEGG(gene = gene_up,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.9,
qvalueCutoff =0.9)
head(kk.up)[,1:6]
dotplot(kk.up)
kk.up=DOSE::setReadable(kk.up, OrgDb='org.Hs.eg.db',
keyType='ENTREZID')#按需替换
tmp = kk.up@result
kk.down <- enrichKEGG(gene = gene_down,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.999,
qvalueCutoff =0.999)
head(kk.down)[,1:6]
dotplot(kk.down)
kk.down=DOSE::setReadable(kk.down, OrgDb='org.Hs.eg.db',
keyType='ENTREZID')#按需替换
kegg_down_dt <- as.data.frame(kk.down)
kegg_up_dt <- as.data.frame(kk.up)
down_kegg<-kegg_down_dt[kegg_down_dt$pvalue<0.01,];down_kegg$group=-1
up_kegg<-kegg_up_dt[kegg_up_dt$pvalue<0.01,];up_kegg$group=1
}
这样的话,会看到上下调基因分开注释就应该有生物学功能的重叠,如下所示:
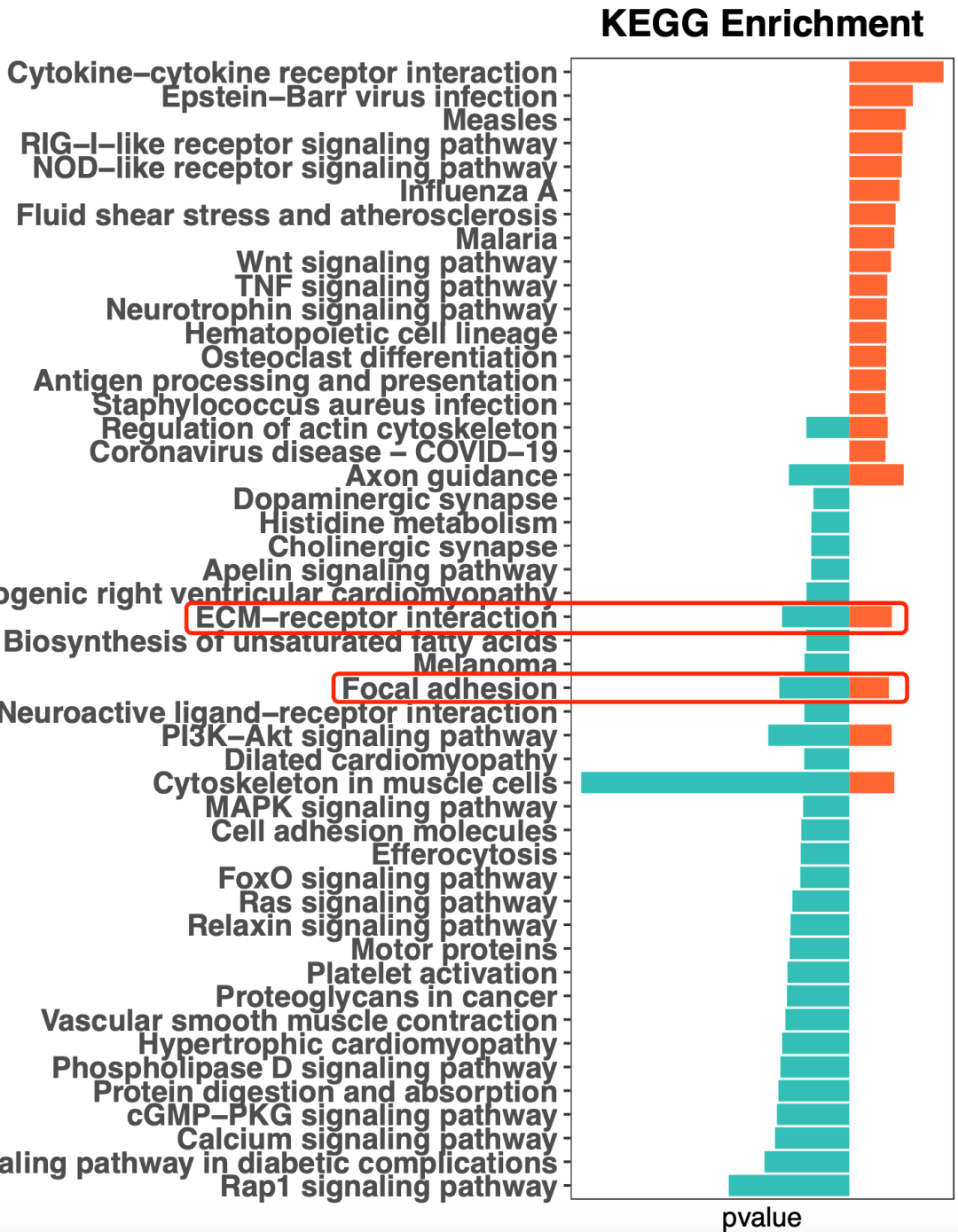
有生物学功能的重叠
实际上,ECM−receptor interaction和Focal adhesion 这两个通路里面的基因是大量重合的,而且 它跟上面的EMT通路也是有大量的重叠,所以我们可以看到作者的emt的gsea结果里面就是左边和右边都有大量的基因 :
如果我们选择GSEA:
就需要采用全部的基因的排序列表去做:
library(gplots)
library(ggplot2)
library(clusterProfiler)
library(GSVA) # BiocManager::install('msigdbr')
library(GSEABase)
library(msigdbr)
load( file = 'DEG_deseq2.Rdata' )
load( file = 'DEG_limma_voom.Rdata' )
load( file = 'DEG_edgeR.Rdata' )
colnames(DEG_deseq2)
colnames(DEG_limma_voom)
geneList = DEG_edgeR$logFC
names(geneList)= rownames(DEG_edgeR)
geneList=sort(geneList,decreasing = T)
head(geneList)
## 1.2 choose hallmark pathway----
all_gene_sets = msigdbr(species = "Homo sapiens", # Homo sapiens or Mus musculus
category = "H" )
length(unique(table(all_gene_sets$gs_name)))
## 1.3 write gsea results to csv----
names(all_gene_sets)
egmt <- GSEA(geneList, TERM2GENE= all_gene_sets[,c('gs_name','gene_symbol')] ,
minGSSize = 1,
pvalueCutoff = 0.99,
verbose=FALSE)
head(egmt)
egmt@result
gsea_results_df <- egmt@result
rownames(gsea_results_df)
可以看到,确实是文章这样的EMT基因列表是下调的:
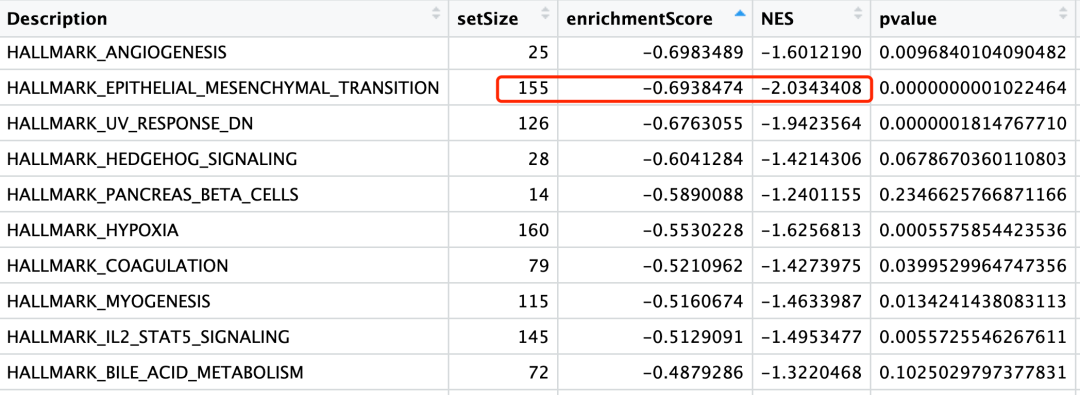
EMT基因列表是下调的
而且可以看具体的gsea细节,如果我们的是先选择统计学显著的上下调基因,然后上下调基因分开注释就应该有生物学功能的重叠,因为它这个通路确实是出现在了我们的排序的两端 :
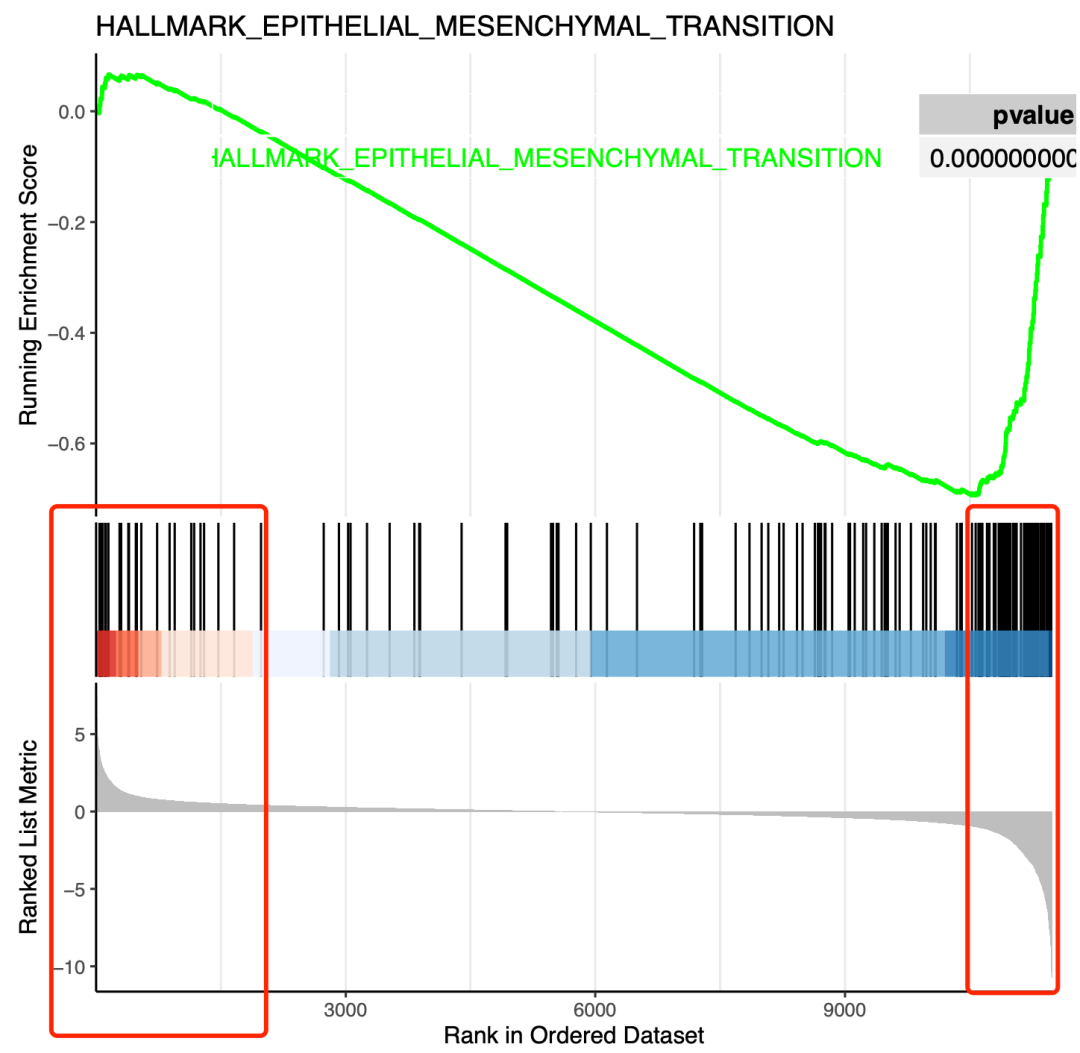
gsea细节
综上所述,上下调基因分开注释就应该有生物学功能的重叠,一切合情合理,如果上下调基因注释到了同一个生物学功能,比如ECM−receptor interaction和Focal adhesion 这两个通路,或者癌症的EMT,其实是应该是具体情况具体分析,因为这个功能本来就是一个大锅饭,emt可以区分为e和m两个完全不一样的,ECM−receptor interaction也是可以拆分成为4个子集。
学徒作业
针对这个数据集进行emt的3个打分,看看能否更进一步的解释这个数据分析的结果!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2024-12-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号