
使用R,将多个卡方关联表测试应用于分组数据帧,并添加一个包含测试p值的新列。
我有一个与下面的示例类似的数据框架(这是我实际数据框架的一个小摘录)。
frequencies <- data.frame(sex=c("female", "female", "male", "male", "female", "female", "male", "male", "female", "female", "male", "male", "female", "female", "male", "male"),
ecotype=c("Crab", "Wave", "Crab", "Wave", "Crab", "Wave", "Crab", "Wave", "Crab", "Wave", "Crab", "Wave", "Crab", "Wave", "Crab", "Wave"),
contig_ID=c("Contig100169_2367", "Contig100169_2367", "Contig100169_2367", "Contig100169_2367", "Contig100169_2367", "Contig100169_2367", "Contig100169_2367", "Contig100169_2367",
"Contig100169_2481", "Contig100169_2481", "Contig100169_2481", "Contig100169_2481", "Contig100169_2481", "Contig100169_2481", "Contig100169_2481", "Contig100169_2481"),
allele=c("p", "p", "p", "p", "q", "q", "q", "q", "p", "p", "p", "p", "q", "q", "q", "q"),
frequency=c(157, 98, 140, 65, 29, 8, 26, 9, 182, 108, 147, 80, 46, 4, 49, 4))
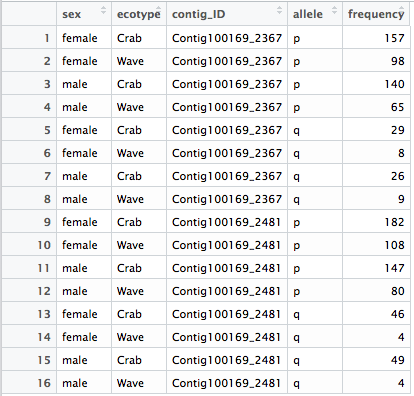
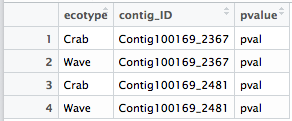
我想对“contig_ID”和“ecotype”的每一个组合分别进行卡方应急测试,测试“性别”和“等位基因”之间的关联。然后,我想在一个表中总结这些结果,其中包括“contig_ID”和“ecotype”的每个组合的p值。例如,从给定的示例表中,我希望得到一个4p值的结果表,如下例所示。
results <- data.frame(ecotype=c("Crab", "Wave", "Crab", "Wave"),
contig_ID=c("Contig100169_2367", "Contig100169_2367", "Contig100169_2481", "Contig100169_2481"),
pvalue=c("pval", "pval", "pval", "pval"))
或者,只向原始表中添加一个p值列也可以,每个组合的p值仅在所有相关行中重复。
我一直试图将lapply()和summarise()等函数与chisq.test()结合起来来实现这一目标,但到目前为止还没有成功。我还试图使用一种类似于此的方法:R chi squared test (3x2 contingency table) for each row in a table,但也无法使其工作。
回答 1
Stack Overflow用户
发布于 2018-04-04 21:04:41
我们可以对contig_ID和ecotype列进行分组,并使用转换成矩阵的数据创建嵌套数据框架,如下所示。
library(tidyverse)
frequencies2 <- frequencies %>%
group_by(contig_ID, ecotype) %>%
nest() %>%
mutate(M = map(data, function(dat){
dat2 <- dat %>% spread(sex, frequency)
M <- as.matrix(dat2[, -1])
row.names(M) <- dat2$allele
return(M)
}))如果我们查看M列的第一个元素,我们将发现来自每个组的数据被转换为一个矩阵。
frequencies2$M[[1]]
# female male
# p 157 140
# q 29 26从这里,我们可以将chisq.test应用于每个矩阵,并提取出p值。frequencies3是最后的输出。
frequencies3 <- frequencies2 %>%
mutate(pvalue = map_dbl(M, ~chisq.test(.x)$p.value)) %>%
select(-data, -M) %>%
ungroup()
frequencies3
# # A tibble: 4 x 3
# contig_ID ecotype pvalue
# <fct> <fct> <dbl>
# 1 Contig100169_2367 Crab 1.00
# 2 Contig100169_2367 Wave 0.434
# 3 Contig100169_2481 Crab 0.284
# 4 Contig100169_2481 Wave 0.958https://stackoverflow.com/questions/49659103
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号