
如何利用entrez.efetch获得特定的蛋白质序列?
如何利用entrez.efetch获得特定的蛋白质序列?
提问于 2013-11-14 19:18:05
我试图通过一个基因id (GI)号从NCBI中获取蛋白质序列,使用Biopython的Entrez.fetch()函数。
proteina = Entrez.efetch(db="protein", id= gi, rettype="gb", retmode="xml").然后,我使用以下方法读取数据:
proteinaXML = Entrez.read(proteina).我可以打印结果,但是我不知道如何单独获得蛋白序列。
一旦结果显示出来,我就可以手动到达蛋白质。或者我使用以下方法检查XML树:
proteinaXML[0]["GBSeq_feature-table"][2]["GBFeature_quals"][6]['GBQualifier_value'].但是,根据提交的蛋白质的GI,XML树可能会有所不同。使这一过程难以可靠地自动化。
我的问题:是否可能只检索蛋白质序列,而不是整个XML?或者,考虑到XML文件的结构可能因蛋白质而异,我如何从XML文件中提取蛋白质序列?
谢谢
回答 1
Stack Overflow用户
回答已采纳
发布于 2013-11-25 17:52:11
好的一点是,XML中的数据库条目在不同作者提交的蛋白质中确实有所不同。
I已经制定了一种算法来“搜索”来自XML树的蛋白质序列:
import os
import sys
from Bio import Entrez
from Bio import SeqIO
gi = '1293613' # example gene id
Entrez.email= "you@email.com" # Always tell NCBI who you are
protina = Entrez.efetch(db="protein", id=gi, retmode="xml") # fetch the xml
protinaXML = Entrez.read(protina)[0]
seqs = [] # store candidate protein seqs
def seqScan(xml): # recursively collect protein seqs
if str(type(xml))=="<class 'Bio.Entrez.Parser.ListElement'>":
for ele in xml:
seqScan(ele)
elif str(type(xml))=="<class 'Bio.Entrez.Parser.DictionaryElement'>":
for key in xml.keys():
seqScan(xml[key])
elif str(type(xml))=="<class 'Bio.Entrez.Parser.StringElement'>":
# v___THIS IS THE KEYWORD FILTER_____v
if (xml.startswith('M') and len(xml))>10: # 1) all proteins start with M (methionine)
seqs.append(xml) # 2) filters out authors starting with M
seqScan(protinaXML) # run the recursive sequence collection
print(seqs) # print the goods!注意:在罕见的情况下(取决于“关键字筛选器”),它可能幽默地抓取不想要的字符串,比如作者名,以'M‘开头,其缩写名称超过10个字符长(下图):
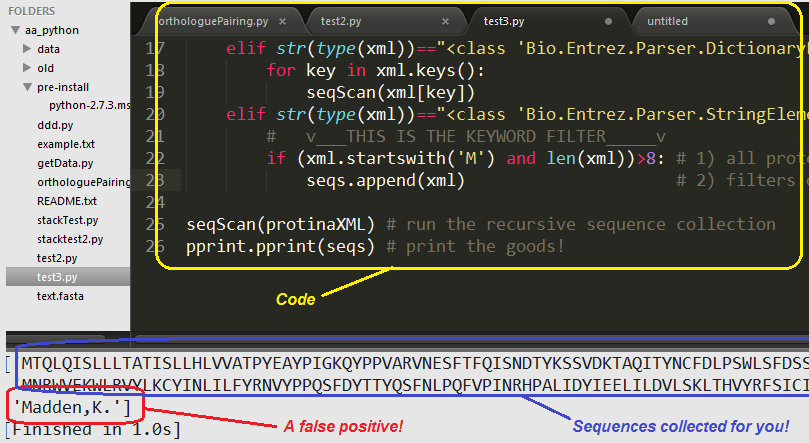
希望这能帮上忙!
页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/19986397
复制相关文章
相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号