
核酸词的系统发育树
核酸词的系统发育树
提问于 2021-01-14 16:30:57
如果对n个核酸序列构造了一个词频表( ATG对应于长度为2,AT和TG的两个单词),则该表可用于(直接或经过PCA降维后)计算这些序列的距离矩阵,然后将其聚为一个系统发育树(doi:10.1007/s 00285-002-0185-3):
library(sequinr)
Bat1 <- read.fasta(file="bat1.FASTA")
Bat1.seq <- Bat1[[1]]
Bat1.count <- as.vector(count(Bat1.seq, 2)) # count word frequencies, k < log4(Sequence length)
...
Counts <- rbind(Bat1.count, ...)
rownames(Counts) <- c("Bat1", ...)
colnames(Counts) <- c(rownames(count(Bat1.seq, 2)))
RowCounts <- rowSums(Counts)
Counts.norm <- Counts/RowCounts # normalise word counts for different sequence length
distance <- dist(Counts.norm, method = "euclidian")
hc <- hclust(distance, method = "average")
plot(hc)几种病毒序列的系统发育树
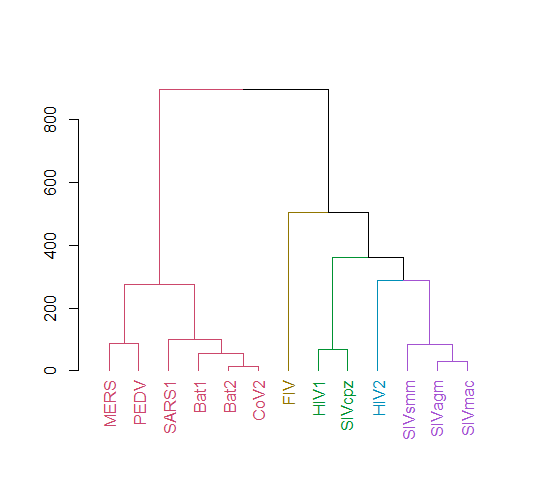
这是令人惊讶的好,输出看起来类似于一棵树获得的多序列对齐与ClustalX,但计算时间是秒,而不是小时。
问:如何衡量这些树的质量,选择最优的单词长度k或(如果使用PCA )的最优分量q,以及距离和聚类方法?最好不要对随机序列进行冗长的引导;-)。
回答 1
Stack Overflow用户
发布于 2021-01-21 10:57:14
这棵树最重要的特点是它是,而不是系统发育的!
在系统发育过程中,边缘反映了进化过程,我们问两个分类群是否有一个共同的祖先,这有多大的可能性。相反,OP图像中的树状图代表了类群之间的DNA序列组成相似性,因此是一种物化树。在决定是否使用所建议的方法时,理解系统发育和物候树之间的区别是至关重要的。如果测试的目的是推断病毒之间的进化关系,那么这种方法是不合适的。
由于树不是一个系统发育,这种关系不需要用进化史的意义来检验。
页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/65722923
复制相关文章
相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号