
如何在使用多变量对象或两个单独对象的组合ggplot2图的基础上添加附加统计信息
我有一个ggplot2图,它将两个单独的小提琴情节绘制到一个图上,这个例子给出了这个图(感谢@jared_mamrot提供它):
library(tidyverse)
data("Puromycin")
head(Puromycin)
dat1 <- Puromycin %>%
filter(state == "treated")
dat2 <- Puromycin %>%
filter(state == "untreated")
mycp <- ggplot() +
geom_violin(data = dat1, aes(x= state, y = conc, colour = "Puromycin (Treatment1)")) +
geom_violin(data = dat2, aes(x= state, y = conc, colour = "Puromycin (Treatment2)"))
mycp
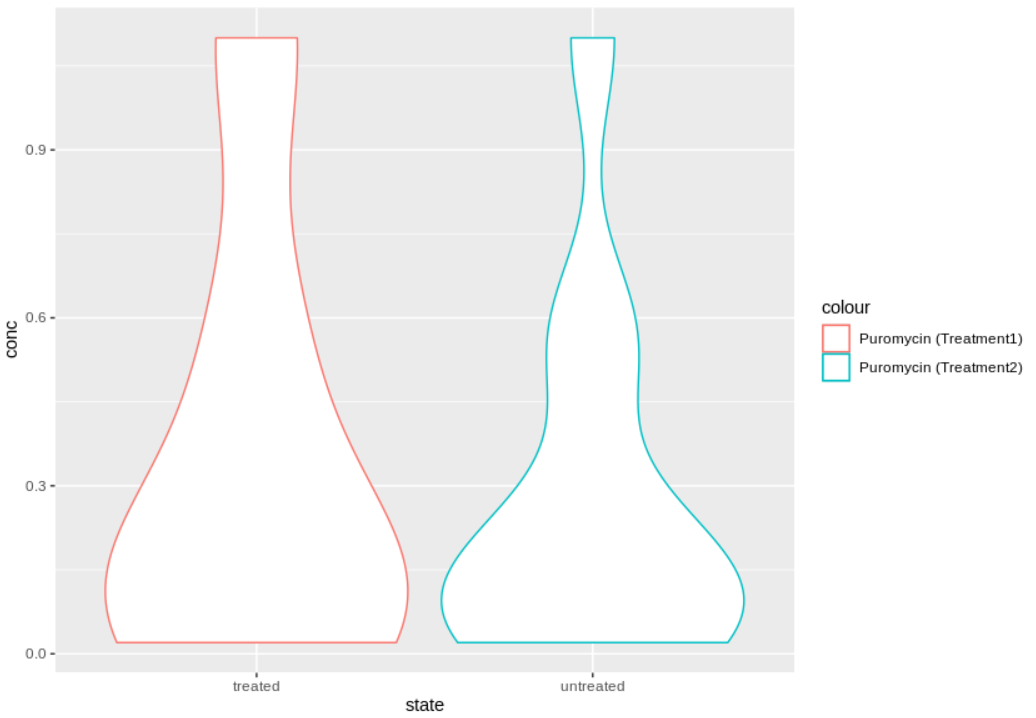
我想添加一个方格图或其他汇总统计信息,如http://www.sthda.com/english/wiki/ggplot2-violin-plot-quick-start-guide-r-software-and-data-visualization和https://www.maths.usyd.edu.au/u/UG/SM/STAT3022/r/current/Misc/data-visualization-2.1.pdf中的统计数据,但是尝试这些地方所建议的代码并不会更改原始图。
mycp + geom_boxplot()谢谢你的阅读,希望这是有意义的!
========================================================================== 更新
所以上面的例子并不能准确地反映我现在的情况。本质上,我想将统计数据应用到一个组合的ggplot2图上,它使用两个单独的对象作为其变量(这里是TNBC_List1和ER_List1) --这里有一个示例(很抱歉,更长的示例是,我在创建一个简单的可复制示例时遇到了困难,而且我一般对编码非常陌生):
# Libraries -------------------------------------------------------------
library(BiocManager)
library(GEOquery)
library(plyr)
library(dplyr)
library(Matrix)
library(devtools)
library(Seurat)
library(ggplot2)
library(cowplot)
library(SAVER)
library(metap)
library(multtest)
# Loading Raw Data into RStudio ----------------------------------
filePaths = getGEOSuppFiles("GSE75688")
tarF <- list.files(path = "./GSE75688/", pattern = "*.tar", full.names = TRUE)
tarF
untar(tarF, exdir = "./GSE75688/")
gzipF <- list.files(path = "./GSE75688/", pattern = "*.gz", full.names = TRUE)
ldply(.data = gzipF, .fun = gunzip)
list.files(path = "./GSE75688/", full.names = TRUE)
list.files(path = "./GSE75688/", pattern = "\\.txt$",full.names = TRUE)
# full matrix ----------------------------------------------------------
fullmat <- read.table(file = './GSE75688//GSE75688_GEO_processed_Breast_Cancer_raw_TPM_matrix.txt',
sep = '\t', header = FALSE, stringsAsFactors = FALSE)
fullmat <- data.frame(fullmat[,-1], row.names=fullmat[,1])
colnames(fullmat) <- as.character(fullmat[1, ])
fullmat <- fullmat[-1,]
fullmat <- as.matrix(fullmat)
# BC01 ER+ matrix -----------------------------------------------------------
BC01mat <- grep(pattern =c("^BC01") , x = colnames(fullmat), value = TRUE)
BC01mat = fullmat[,grepl(c("^BC01"),colnames(fullmat))]
BC01mat = BC01mat[,!grepl("^BC01_Pooled",colnames(BC01mat))]
BC01mat = BC01mat[,!grepl("^BC01_Tumor",colnames(BC01mat))]
BC01pdat <- data.frame("samples" = colnames(BC01mat), "treatment" = "ER+")
# BC07 TNBC matrix -----------------------------------------------------------
BC07mat <- grep(pattern =c("^BC07") , x = colnames(fullmat), value = TRUE)
BC07mat <- fullmat[,grepl(c("^BC07"),colnames(fullmat))]
BC07mat <- BC07mat[,!grepl("^BC07_Pooled",colnames(BC07mat))]
BC07mat <- BC07mat[,!grepl("^BC07_Tumor",colnames(BC07mat))]
BC07mat <- BC07mat[,!grepl("^BC07LN_Pooled",colnames(BC07mat))]
BC07mat <- BC07mat[,!grepl("^BC07LN",colnames(BC07mat))]
BC07pdat <- data.frame("samples" = colnames(BC07mat), "treatment" = "TNBC")
#merge samples together =========================================================================
joined <- cbind(BC01mat, BC07mat)
pdat_joined <- rbind(BC01pdat, BC07pdat)
#fdat ___________________________________________________________________________________
fdat <- grep(pattern =c("gene_name|gene_type") , x = colnames(fullmat), value = TRUE)
fdat <- fullmat[,grepl(c("gene_name|gene_type"),colnames(fullmat))]
fdat <- as.data.frame(fdat, stringsAsFactors = FALSE)
fdat <- setNames(cbind(rownames(fdat), fdat, row.names = NULL),
c("ensembl_id", "gene_short_name", "gene_type"))
rownames(pdat_joined) <- pdat_joined$samples
rownames(fdat) = make.names(fdat$gene_short_name, unique=TRUE)
rownames(joined) <- rownames(fdat)
# Create Seurat Object __________________________________________________________________
joined <- as.data.frame(joined)
sobj_pre <- CreateSeuratObject(counts = joined)
sobj_pre <-AddMetaData(sobj_pre,metadata=pdat_joined)
head(sobj_pre@meta.data)
#gene name input
sobj_pre[["RNA"]]@meta.features<-fdat
head(sobj_pre[["RNA"]]@meta.features)
#Downstream analysis -------------------------------------------------------
sobj <- sobj_pre
sobj <- FindVariableFeatures(object = sobj, mean.function = ExpMean, dispersion.function = LogVMR, nfeatures = 2000)
sobj <- ScaleData(object = sobj, features = rownames(sobj), block.size = 2000)
sobj <- RunPCA(sobj, npcs = 100, ndims.print = 1:10, nfeatures.print = 5)
sobj <- FindNeighbors(sobj, reduction = "pca", dims = 1:4, nn.eps = 0.5)
sobj <- FindClusters(sobj, resolution = 1, n.start = 10)
umap.method = 'umap-learn'
metric = 'correlation'
sobj <- RunUMAP(object = sobj, reduction = "pca", dims = 1:4,min.dist = 0.5, seed.use = 123)
p0 <- DimPlot(sobj, reduction = "umap", pt.size = 0.1,label=TRUE) + ggtitle(label = "Title")
p0
# ER+ score computation -------------------
ERlist <- list(c("CPB1", "RP11-53O19.1", "TFF1", "MB", "ANKRD30B",
"LINC00173", "DSCAM-AS1", "IGHG1", "SERPINA5", "ESR1",
"ILRP2", "IGLC3", "CA12", "RP11-64B16.2", "SLC7A2",
"AFF3", "IGFBP4", "GSTM3", "ANKRD30A", "GSTT1", "GSTM1",
"AC026806.2", "C19ORF33", "STC2", "HSPB8", "RPL29P11",
"FBP1", "AGR3", "TCEAL1", "CYP4B1", "SYT1", "COX6C",
"MT1E", "SYTL2", "THSD4", "IFI6", "K1AA1467", "SLC39A6",
"ABCD3", "SERPINA3", "DEGS2", "ERLIN2", "HEBP1", "BCL2",
"TCEAL3", "PPT1", "SLC7A8", "RP11-96D1.10", "H4C8",
"PI15", "PLPP5", "PLAAT4", "GALNT6", "IL6ST", "MYC",
"BST2", "RP11-658F2.8", "MRPS30", "MAPT", "AMFR", "TCEAL4",
"MED13L", "ISG15", "NDUFC2", "TIMP3", "RP13-39P12.3", "PARD68"))
sobj <- AddModuleScore(object = sobj, features = ERlist, name = "ER_List")
#TNBC computation -------------------
tnbclist <- list(c("FABP7", "TSPAN8", "CYP4Z1", "HOXA10", "CLDN1",
"TMSB15A", "C10ORF10", "TRPV6", "HOXA9", "ATP13A4",
"GLYATL2", "RP11-48O20.4", "DYRK3", "MUCL1", "ID4", "FGFR2",
"SHOX2", "Z83851.1", "CD82", "COL6A1", "KRT23", "GCHFR",
"PRICKLE1", "GCNT2", "KHDRBS3", "SIPA1L2", "LMO4", "TFAP2B",
"SLC43A3", "FURIN", "ELF5", "C1ORF116", "ADD3", "EFNA3",
"EFCAB4A", "LTF", "LRRC31", "ARL4C", "GPNMB", "VIM",
"SDR16C5", "RHOV", "PXDC1", "MALL", "YAP1", "A2ML1",
"RP1-257A7.5", "RP11-353N4.6", "ZBTB18", "CTD-2314B22.3", "GALNT3",
"BCL11A", "CXADR", "SSFA2", "ADM", "GUCY1A3", "GSTP1",
"ADCK3", "SLC25A37", "SFRP1", "PRNP", "DEGS1", "RP11-110G21.2",
"AL589743.1", "ATF3", "SIVA1", "TACSTD2", "HEBP2"))
sobj <- AddModuleScore(object = sobj, features = tnbclist, name = "TNBC_List")
#ggplot2 issue ----------------------------------------------------------------------------
sobj[["ClusterName"]] <- Idents(object = sobj)
sobjlists <- FetchData(object = sobj, vars = c("ER_List1", "TNBC_List1", "ClusterName"))
library(reshape2)
melt(sobjlists, id.vars = c("ER_List1", "TNBC_List1", "ClusterName"))
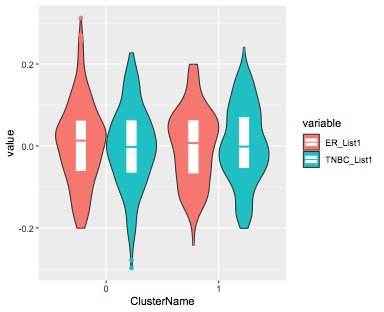
p <- ggplot() + geom_violin(data = sobjlists, aes(x= ClusterName, y = ER_List1, fill = ER_List1, colour = "ER+ Signature"))+ geom_violin(data = sobjlists, aes(x= ClusterName, y = TNBC_List1, fill = TNBC_List1, colour="TNBC Signature"))====================================================================== 扩展
如果您想这样做,但是要使用两个对象(例如,sobjlists1和sobjlists2 ),而不是我的示例显示的对象(两个变量,但一个对象),则rbind两个对象,然后执行@StupidWolf所说的操作
library(reshape2)
sobjlists1= melt(sobjlists1, id.vars = "treatment")
sobjlists2= melt(sobjlists2, id.vars = "treatment")
combosobjlists <- rbind(sobjlists1, sobjlists2)然后继续使用combosobjlists进行代码处理。
ggplot(combosobjlists,aes(x= ClusterName, y = value)) +
geom_violin(aes(fill=variable)) +
geom_boxplot(aes(col=variable),
width = 0.2,position=position_dodge(0.9))希望这条线能帮上忙!
回答 2
Stack Overflow用户
发布于 2021-06-19 13:53:54
尽量只包含显示问题的最小代码。就像在您的示例中一样,不需要从整个seurat处理开始。您只需向data.frame提供dput(),我们就可以看到ggplot2的问题所在,参见this post。
创建一些示例数据:
library(Seurat)
library(ggplot2)
genes = c(unlist(c(ERlist,tnbclist)))
mat = matrix(rnbinom(500*length(genes),mu=500,size=1),ncol=500)
rownames(mat) = genes
colnames(mat) = paste0("cell",1:500)
sobj = CreateSeuratObject(mat)
sobj = NormalizeData(sobj)添加一些组成的集群:
sobj$ClusterName = factor(sample(0:1,ncol(sobj),replace=TRUE))加上你的模块分数:
sobj = AddModuleScore(object = sobj, features = tnbclist,
name = "TNBC_List",ctrl=5)
sobj = AddModuleScore(object = sobj, features = ERlist,
name = "ER_List",ctrl=5) 我们得到了数据,你需要做的是正确地把它转长。用ggplot2绘制两次图会导致各种问题:
sobjlists = FetchData(object = sobj, vars = c("ER_List1", "TNBC_List1", "ClusterName"))
head(sobjlists)
ER_List1 TNBC_List1 ClusterName
cell1 -0.05391108 -0.008736057 1
cell2 0.07074816 -0.039064126 1
cell3 0.08688374 -0.066967324 1
cell4 -0.12503649 0.120665057 0
cell5 0.05356685 -0.072293651 0
cell6 -0.20053804 0.178977042 1应该是这样的:
library(reshape2)
sobjlists = melt(sobjlists, id.vars = "ClusterName")
ClusterName variable value
1 1 ER_List1 -0.05391108
2 1 ER_List1 0.07074816
3 1 ER_List1 0.08688374
4 0 ER_List1 -0.12503649
5 0 ER_List1 0.05356685
6 1 ER_List1 -0.20053804现在我们来策划:
ggplot(sobjlists,aes(x= ClusterName, y = value)) +
geom_violin(aes(fill=variable)) +
geom_boxplot(aes(col=variable),
width = 0.2,position=position_dodge(0.9))
Stack Overflow用户
发布于 2021-06-17 20:43:00
为了能够在不指定数据的情况下使用绘图中的数据(如geom_boxplot() ),需要将数据放在ggplot()函数调用中。然后,以下函数可以继承它们。
你也不需要额外的小提琴每种颜色的情节
library(tidyverse)
data("Puromycin")
head(Puromycin)
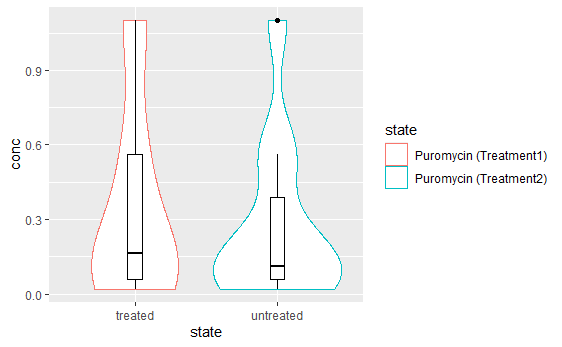
mycp <- ggplot(Puromycin,aes(x= state, y = conc, colour=state))+geom_violin()
mycp + geom_boxplot(width=0.1, color= "black") +
scale_color_discrete(
labels= c("Puromycin (Treatment1)","Puromycin (Treatment2)")
)结果:
https://stackoverflow.com/questions/68025965
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号