
与VMD的psfgen模块混淆
总的来说,我对VMD和编程非常陌生。我需要将两个子单元的pdb文件合并成两个子单元的组合pdb和psf文件。我使用了Namd教程,使用了两个名为BChain270VerCTrue.pdb和barn_noH2o_ChainD.pdb的pdb文件,并在VMD中运行了这个pgn:
package require psfgen
topology top_all27_prot_lipid.inp
pdbalias residue HIS HSE
pdbalias atom ILE CD1 CD
segment A {pdb BChain270VerCTrue.pdb}
segment D {pdb barn_noH2o_ChainD.pdb}
coordpdb BChain270VerCTrue.pdb A
coordpdb barn_noH2o_ChainD.pdb D
guesscoord
writepdb BarnaseBarnstar270True.pdb
writepsf BarnaseBarnstar270True.psf然而,这产生了一种损坏的pdb,它有两个子单元共价结合。我怎么才能解决这个问题?
当仅在VMD中单独使用时,这两个pdb文件的外观如下:
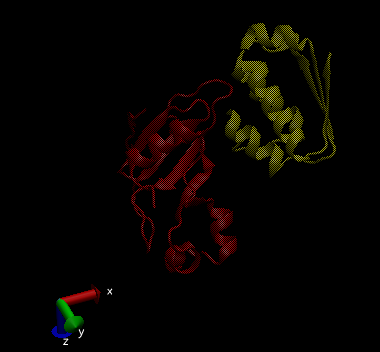
这就是代码所显示的内容:
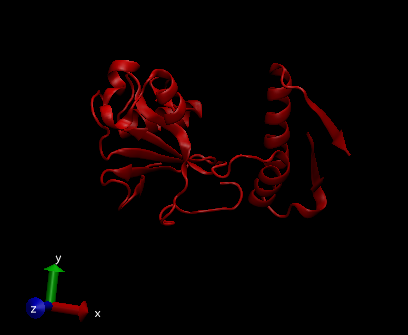
回答 1
Stack Overflow用户
发布于 2022-04-20 07:10:05
巴那酶和巴星是淀粉芽孢杆菌产生的胞外核糖核酸酶及其胞内抑制剂。这两种蛋白质具有很强的结合能,因此常用于研究蛋白质与蛋白质的相互作用.为了模拟硅中的这种相互作用,我们需要一个适当的强迫场,2)拓扑文件,3)分子动力学软件包,如NAMD、Gromacs或Amber。另外,对PDB文件格式和结构生物学以及Python或Bash脚本技巧的了解也很少。
要生成一个拓扑文件,必须分别生成每个组件,如果PDB文件有两个链,那么其中一个必须将它们分开。然而,当我们必须将它们放回时,我们必须遵循PDB文件格式规则。(详见https://www.wwpdb.org/documentation/file-format)。
PDB格式由许多列组成,但第一列包含原子信息,如ATOM、HETATOM或TER。最后一个三指定一个链的终止,没有它,一些软件认为他们是一个单链,就像你的情况。
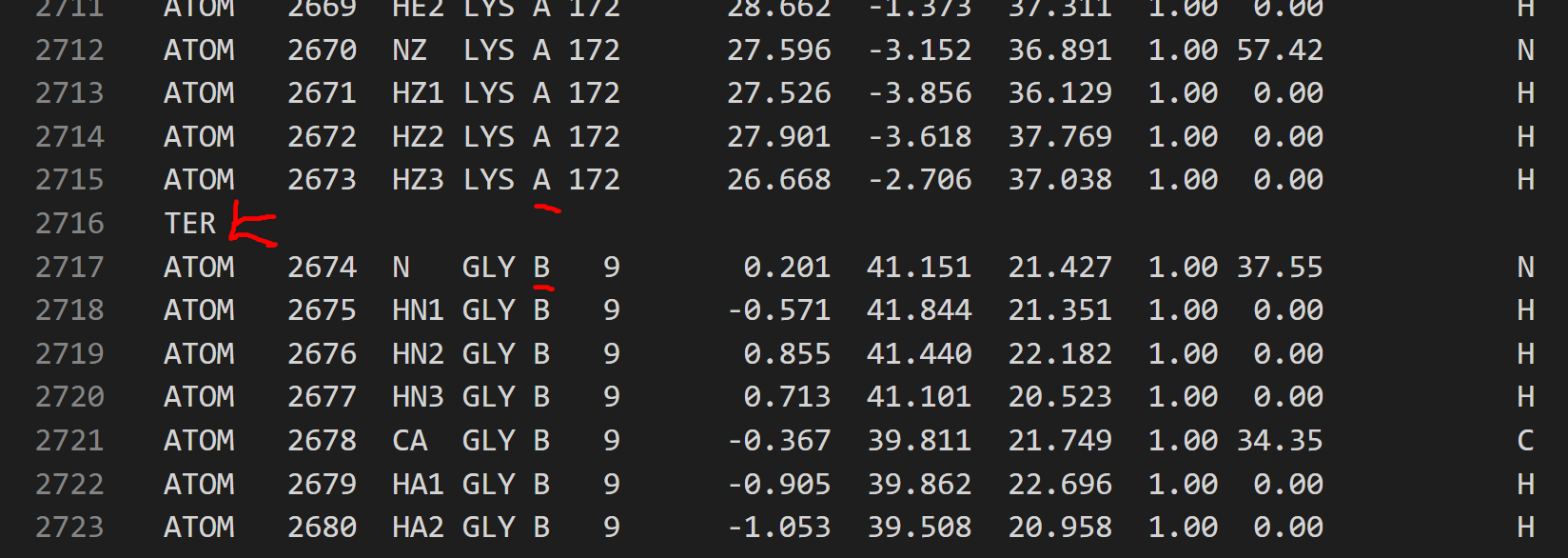
确保两条链分开。您可以使用一个简单的文本编辑器来放置。
https://stackoverflow.com/questions/69653794
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号