Cell | 空间转录组揭示胰腺癌恶性转化的关键细胞状态与微环境重塑机制

Cell | 空间转录组揭示胰腺癌恶性转化的关键细胞状态与微环境重塑机制

生信大杂烩
发布于 2026-05-08 19:45:41
发布于 2026-05-08 19:45:41
导读
胰腺导管腺癌(PDAC)是致死率最高的恶性肿瘤之一,5年生存率不足12%。这种极端的预后,很大程度上源于其隐匿的早期进展——当我们能够检测到它时,往往已经是晚期。良性病变如何跨越那道"不可逆"的门槛,演变为浸润性癌? 这一问题困扰肿瘤学界数十年。
2026年5月,来自纪念斯隆-凯特琳癌症中心(MSKCC)Dana Pe'er 和 Scott W. Lowe 实验室的研究团队在 Cell 杂志发表了重磅研究。他们整合单细胞转录组学与空间转录组学,在胰腺癌小鼠模型中精准捕捉到良恶性转化的关键节点——一群罕见的祖细胞样细胞(progenitor-like cells),并揭示这群细胞如何在促癌与抑癌力量的拉锯中,构建出一个自我强化的"癌样生态位"(cancer-like niche),最终决定肿瘤是否发生。
一、研究背景:被忽视的"中间地带"
在胰腺癌的发生发展模型中,Kras 突变是最早的驱动事件——它在胰腺上皮中广泛存在,并可诱导胰腺上皮内瘤变(PanIN)等癌前病变。但仅有Kras突变并不足以致癌,还需要 TP53、CDKN2A、SMAD4 等肿瘤抑制基因的失活,才能真正跨越良恶性转化的门槛。
然而,这些肿瘤抑制因子具体在哪些细胞、哪个时间窗口发挥作用,始终是个黑箱。研究团队此前的工作已提示,在Kras突变背景下,组织损伤(如胰腺炎)可诱导一类特殊的祖细胞样细胞出现——它们高表达间质祖细胞标志物(Nes、Msn、Hmga2、Vim),在转录上最接近胰腺癌,具有高度可塑性。但这群细胞与肿瘤抑制机制、微环境重塑之间的关系从未被系统阐明。
本研究的核心问题因此被提炼为:促癌力量(致癌KRAS信号)与抑癌力量(p53、CDKN2A、SMAD4)如何在同一群细胞中相遇?它们的博弈如何决定良恶性转化的最终走向?
二、研究体系:用荧光报告系统"捕捉"p53丢失的瞬间
为了捕捉自发性p53失活这一关键事件,研究团队使用了精心设计的 KPLOH 小鼠模型:
- 该模型来源于经典KPC小鼠(KrasG12D; Trp53flox/+; Ptf1a-Cre),并整合荧光报告基因
- mKate2+/GFP+ 双阳性细胞:表示野生型p53完整的Kras突变上皮细胞
- mKate2+/GFP− 单阳性细胞:GFP与野生型p53等位基因共缺失,标记自发p53杂合性缺失(LOH)的细胞
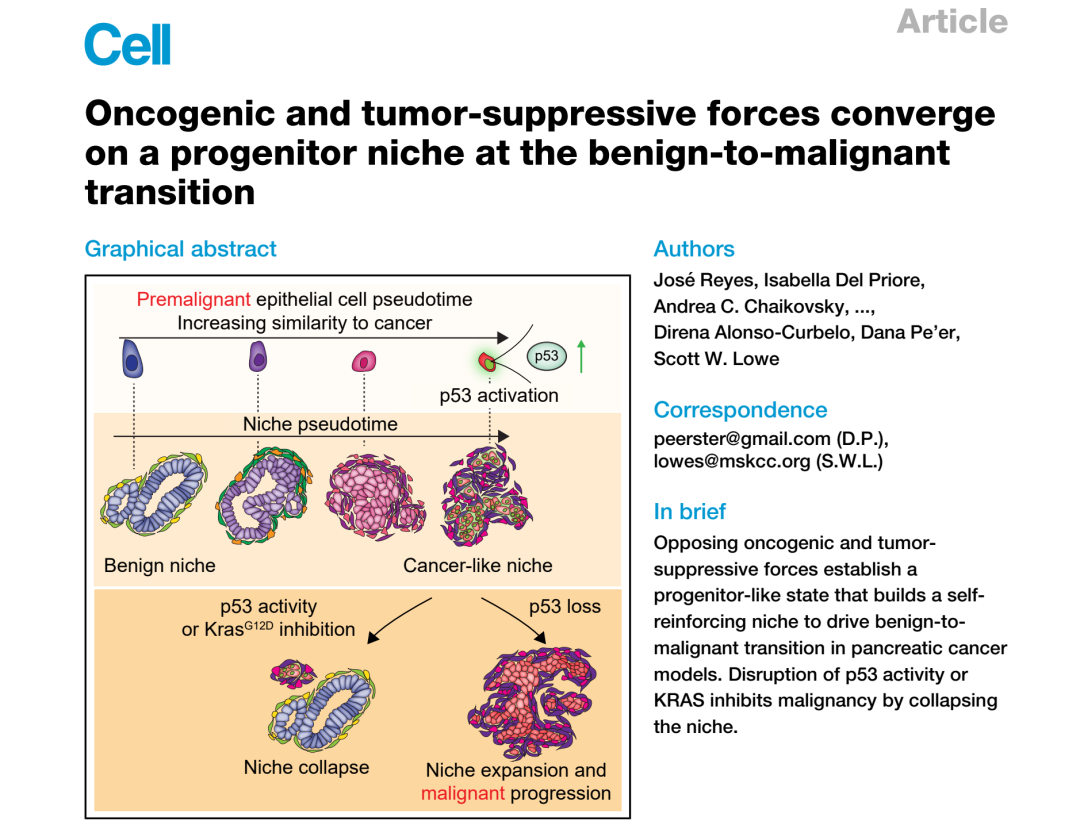
▲ 图1 | KPLOH小鼠模型设计(A-B)及单细胞转录组全景(D-L)。在肿瘤前期,p53缺失细胞(mKate2+/GFP−)极为罕见(1%-3%),但祖细胞样亚群(Progenitor-like)在转录上最接近PDAC(F)。smFISH验证(H)显示祖细胞样细胞(Msn+)在癌前腺体中分散分布,各自独立出现(I-J)。
利用单细胞RNA测序(scRNA-seq),研究者对比了肿瘤前期p53完整细胞、p53缺失细胞以及PDAC来源细胞的转录组,构建了一幅完整的癌前-癌变转录图谱。
关键发现之一:在所有癌前细胞亚群中,祖细胞样细胞在转录上与癌细胞的扩散距离最近,提示其为良恶性转化的直接前体。
三、核心发现:促癌与抑癌程序在祖细胞样亚群中同时"点火"
这一部分是全文最令人震惊的发现之一。
研究者对每个癌前细胞亚群分别比较了p53完整与p53缺失状态下的p53靶基因表达情况。结果令人意外:在大多数亚群中,p53靶基因几乎没有变化——唯独在祖细胞样细胞中,p53靶基因高度激活,并在p53缺失后显著下调(图1G)。
这意味着:虽然Kras突变遍布整个胰腺上皮,p53的抑癌活性却选择性地聚焦于祖细胞样细胞。
更进一步,研究者发现,祖细胞样细胞中同时高度激活的不仅是p53通路,还有另外两大PDAC关键抑癌通路:
- CDKN2A(p16INK4A 与 p19ARF 两种剪接异构体均被激活)
- SMAD4/TGF-β 信号通路
与此同时,这群细胞还高表达KRAS信号、糖酵解/Warburg效应、上皮-间质转化(EMT)等典型促癌程序,以及衰老相关转录特征(图1L)。
促癌程序与抑癌程序在同一细胞中同时激活,形成类似"癌基因诱导的衰老"(OIS)的对峙格局。
这一发现将祖细胞样细胞定义为:PDAC起始过程中,促癌与抑癌力量真正交锋的战场。
四、空间转录组学:解析祖细胞生态位的逐步组装
4.1 G-P扩散轴:上皮可塑性的定量坐标
为在组织原位解析这一过程,研究团队利用Xenium原位空间转录组平台(10× Genomics)设计了480基因panel,对多只KC小鼠胰腺炎诱导后2天(n=10)和3周(n=5)的组织进行了深度空间profiling,共覆盖超过350万个细胞。
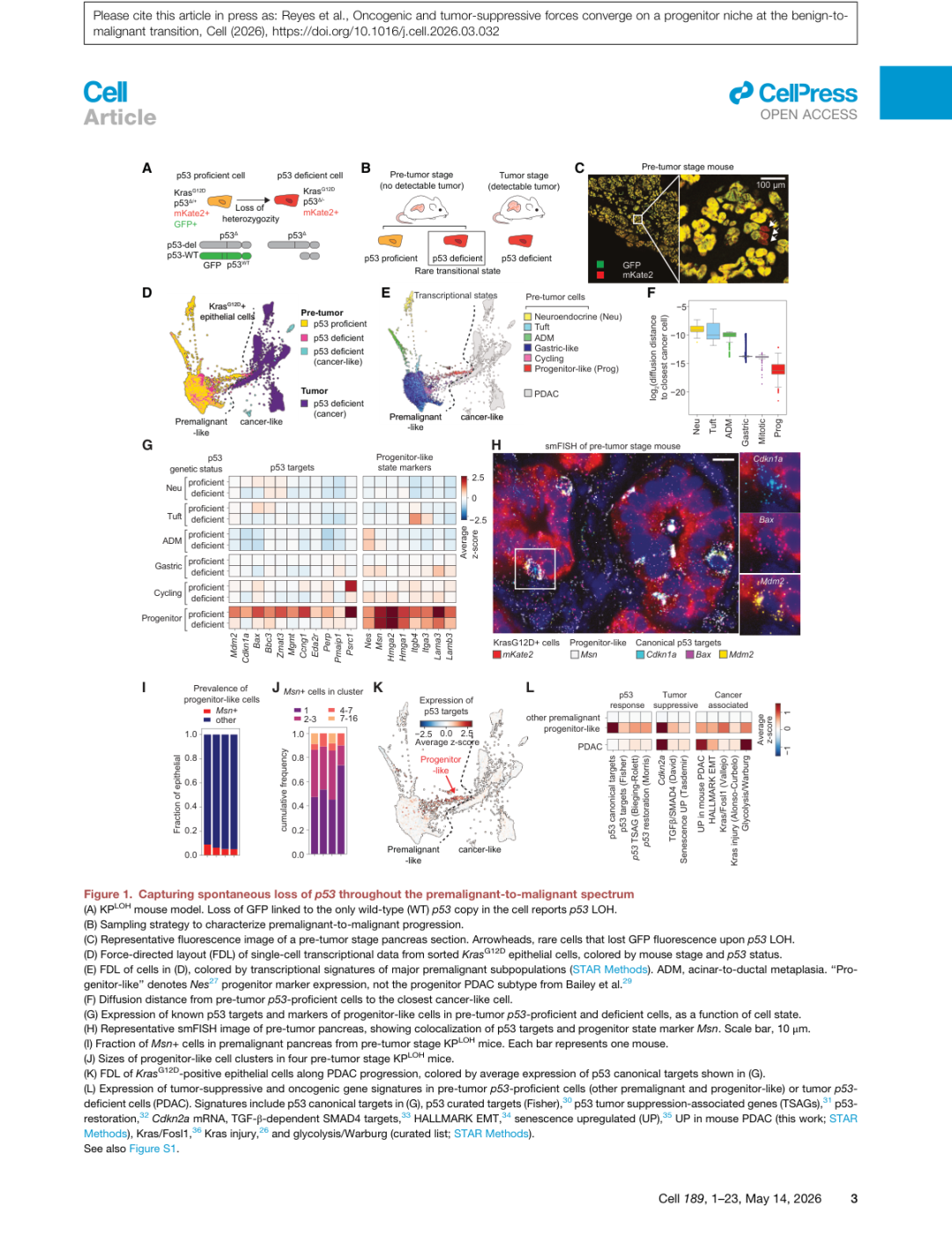
研究者开发了一套整合空间与转录维度的分析框架,通过扩散组分分析(Diffusion Component Analysis)确定了胃样-祖细胞样(G-P)扩散轴——这是上皮细胞转录变异的最主要轴线,依次对应Msn → Hmga2 → Vim的逐步诱导,代表上皮细胞获得祖细胞特征的连续过程。
▲ 图2 | 空间转录组与单细胞embedding(D-E),G-P扩散轴(F)以及沿该轴的组织形态参数变化(H-K):随着细胞向祖细胞样端移动,管腔结构逐渐丧失,上皮密度下降,间质和免疫细胞浸润增加,病灶体积扩大。
4.2 niche pseudotime:从静态切片重建动态演化
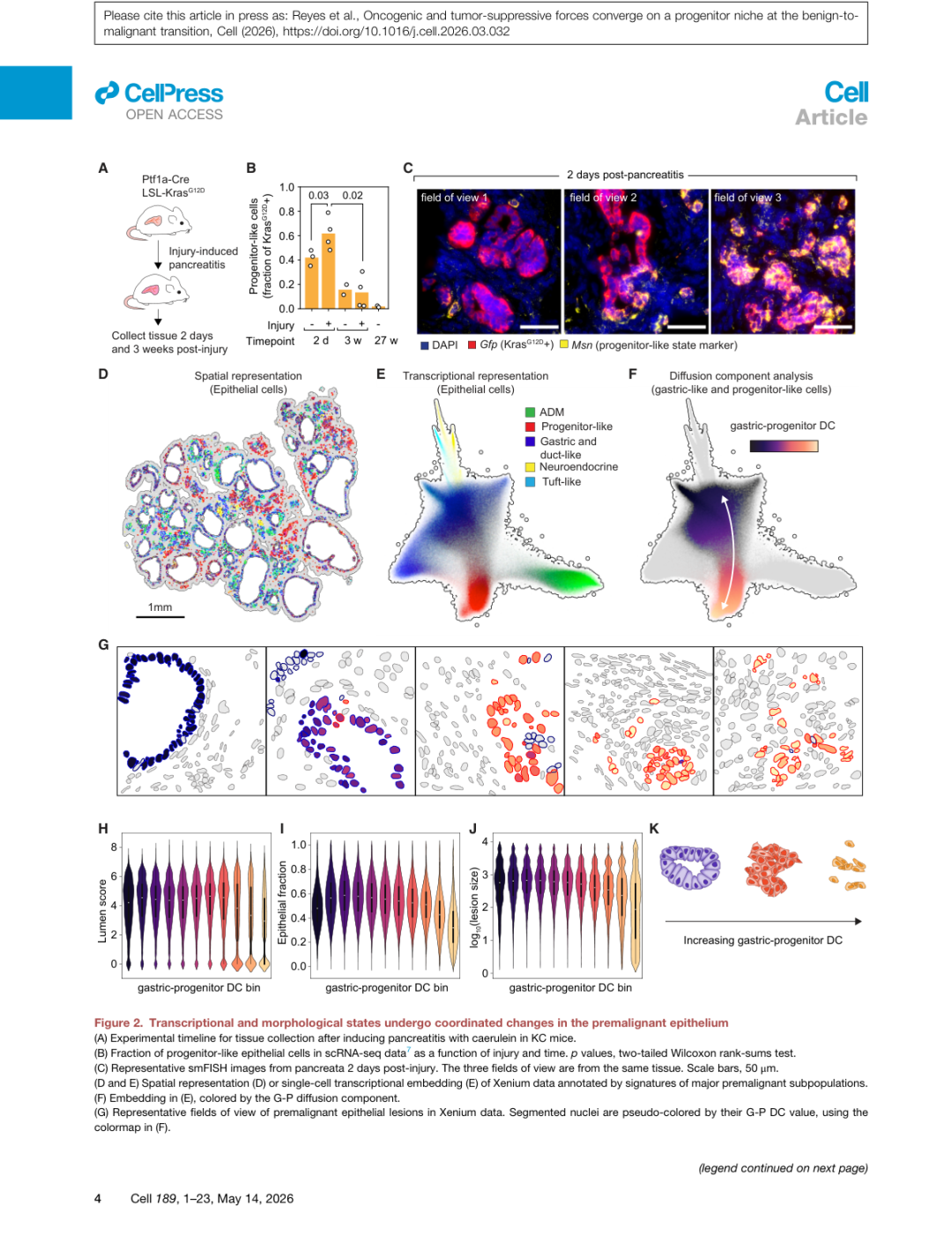
研究者将空间邻域(以上皮细胞为"锚点",半径60μm内的所有细胞构成一个"Niche")按其上皮细胞的平均G-P坐标排序,构建G-P伪时间——从静态组织截面中重建了niche的动态演化轨迹。
▲ 图3 | niche pseudotime分析揭示,随祖细胞特征增加,niche内微环境发生协调重塑:Gli1+ 肌成纤维细胞(myCAF)逐渐被 Tnc+/Ccn2+ myCAF 取代(D-F),免疫髓系细胞从Maf+ TAM转变为Itgax+ 免疫抑制性巨噬细胞(E)。多室的伤口愈合基因特征在祖细胞niche中显著上调(G)。免疫荧光染色(H)证实HMGA2+/uPAR+/ARG1+/TNC+构成祖细胞niche的特征性细胞组合,且在PDAC中高度保守。
这一分析揭示了niche组装的有序进程:
- 早期(胃样niche):周围富含Gli1+肌成纤维细胞和Maf+巨噬细胞
- 中期:随着上皮向祖细胞方向漂移,Tnc+/Postn+/Tgfb1+肌成纤维细胞和Itgax+巨噬细胞逐渐富集
- 晚期(祖细胞niche):形成ARG1+免疫抑制性巨噬细胞 + TNC+活化肌成纤维细胞的"癌样微环境"
这一niche的结构与浸润性PDAC患者的肿瘤微环境高度相似,却出现在肿瘤形成之前——表明癌样niche的构建先于恶性转化。
五、人类验证:慢性胰腺炎患者中存在同源祖细胞niche
为评估上述发现的临床相关性,研究者做了两个层面的人类验证。
转录层面:对67例无癌个体胰腺上皮scRNA-seq数据(Carpenter et al.)投影小鼠祖细胞特征,在ADM/导管样细胞中发现罕见的HMGA1+/MSN+/KRAS信号+/衰老相关程序+细胞亚群,其转录逻辑与小鼠祖细胞高度吻合;同时发现KRT17(基底细胞标志物)是这群细胞的可靠标记物。
空间层面:对67例慢性/急性胰腺炎患者(含27例非肿瘤及40例PDAC瘤旁组织)进行高度多重免疫荧光(IF)分析:
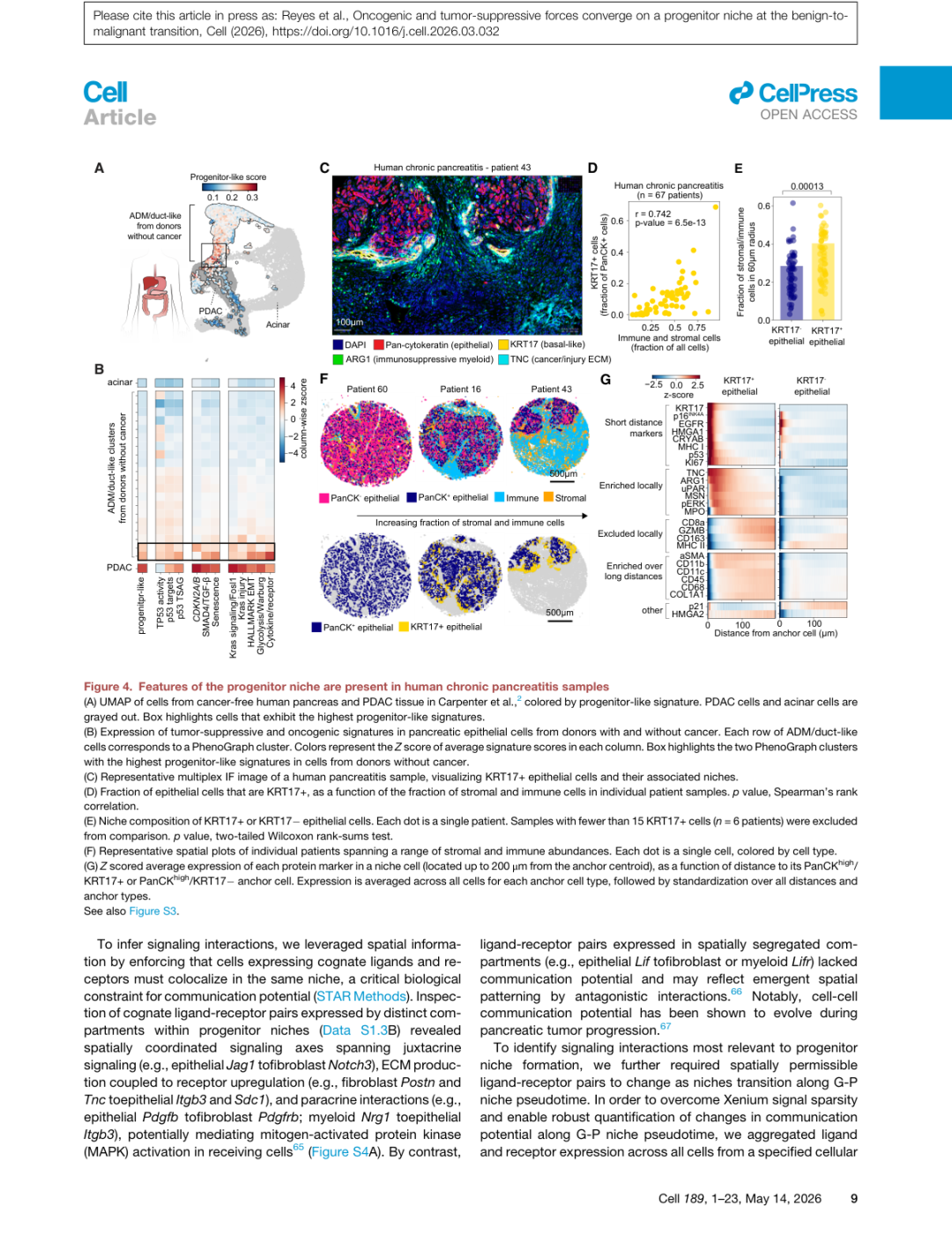
▲ 图4 | 人类慢性胰腺炎样本中,KRT17+上皮细胞的丰度与间质/免疫细胞浸润高度正相关(D,r=0.742,p=6.5e-13)。以KRT17+细胞为中心的同心圆空间分析(G)显示:近端富集p53、p16、HMGA1、ARG1、TNC、uPAR等祖细胞niche标志物,并存在CD8a/GZMB的显著耗竭,提示T细胞排除。
六、通讯电路:祖细胞niche的自我强化机制
niche的形成不是偶然事件,而是多细胞间信号的正反馈回路所驱动。
研究者利用空间约束(要求配体-受体分子必须共定位于同一niche中)推断祖细胞niche中的细胞通讯,发现多条关键信号轴随niche伪时间协调激活:
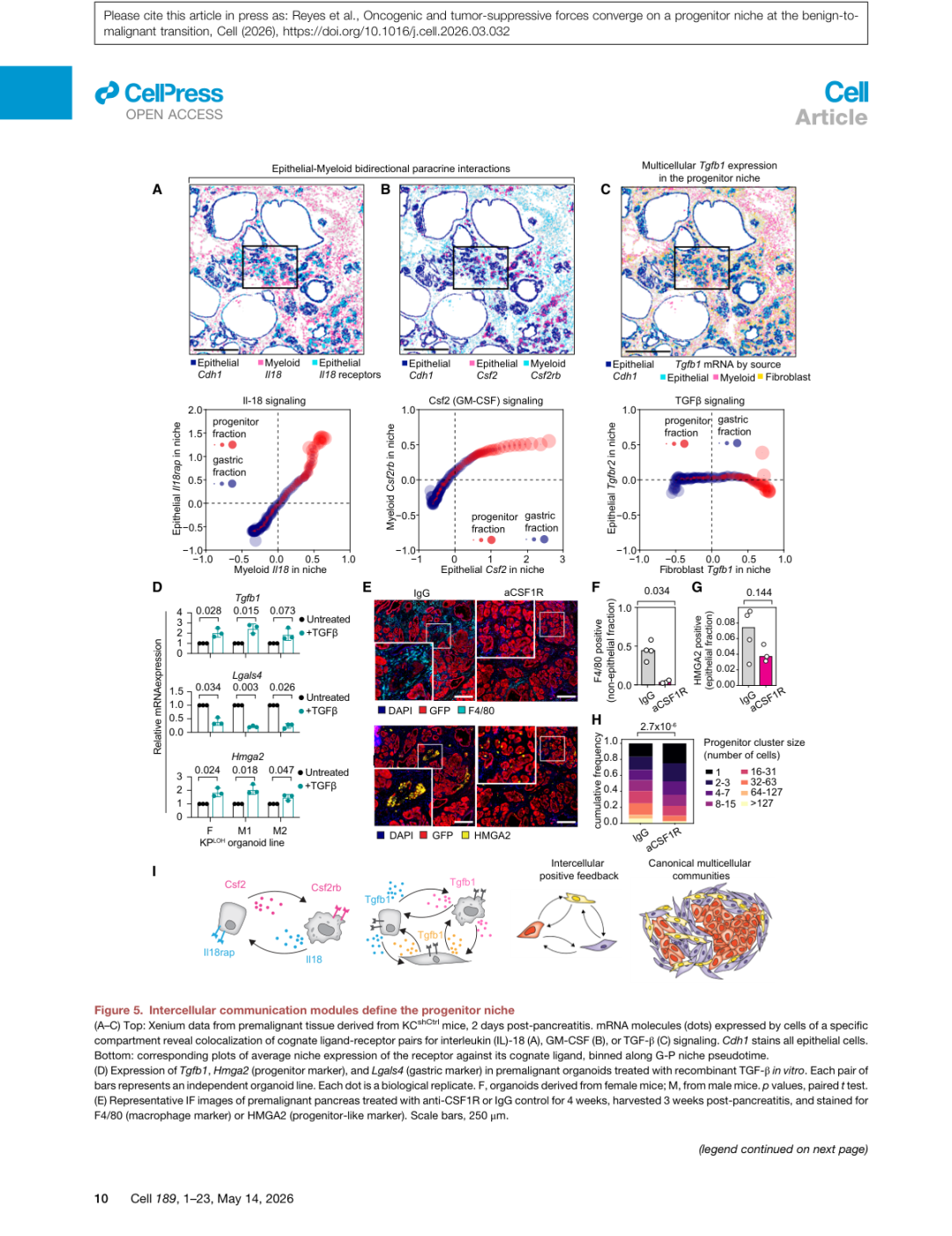
▲ 图5 | 祖细胞niche中的多室信号模块(A-C):上皮-髓系双向 IL-18 信号(A)、GM-CSF/CSF2 信号(B)及跨上皮-成纤维细胞-髓系三室 TGF-β 信号(C)随G-P niche伪时间协调上调,构成正反馈回路。体外TGF-β处理癌前类器官可上调Hmga2并下调胃样标志Lgals4(D),功能验证该通路的作用。anti-CSF1R巨噬细胞清除实验(E-H)证实巨噬细胞对祖细胞niche稳定性和扩张至关重要。
核心通讯模块:
信号轴 | 方向 | 功能意义 |
|---|---|---|
IL-18(髓系→上皮)+ IL-18R(上皮表达) | 双向 | 促进上皮祖细胞状态维持 |
CSF2/GM-CSF(上皮→髓系) | 上皮→巨噬细胞 | 募集和激活免疫抑制性巨噬细胞 |
TGF-β1(多室联合) | 成纤维细胞/髓系→上皮 | 促进上皮间质转化,维持祖细胞特征 |
JAG1-Notch3(上皮→成纤维细胞) | 旁分泌 | 激活肌成纤维细胞 |
功能验证表明:抗CSF1R抗体清除巨噬细胞可显著减少祖细胞样细胞数量并消除大型祖细胞病灶,证明巨噬细胞对niche的稳定性至关重要。
七、靶向干预:阻断KRAS可瓦解祖细胞niche并延缓肿瘤发生
关键问题:如果祖细胞niche是良恶性转化的门户,那么靶向它是否能预防癌症?
研究者在胰腺炎诱导后给予KPLOH小鼠48小时的KRASG12D特异性抑制剂MRTX1133脉冲处理,再用Xenium空间转录组分析其微环境变化。
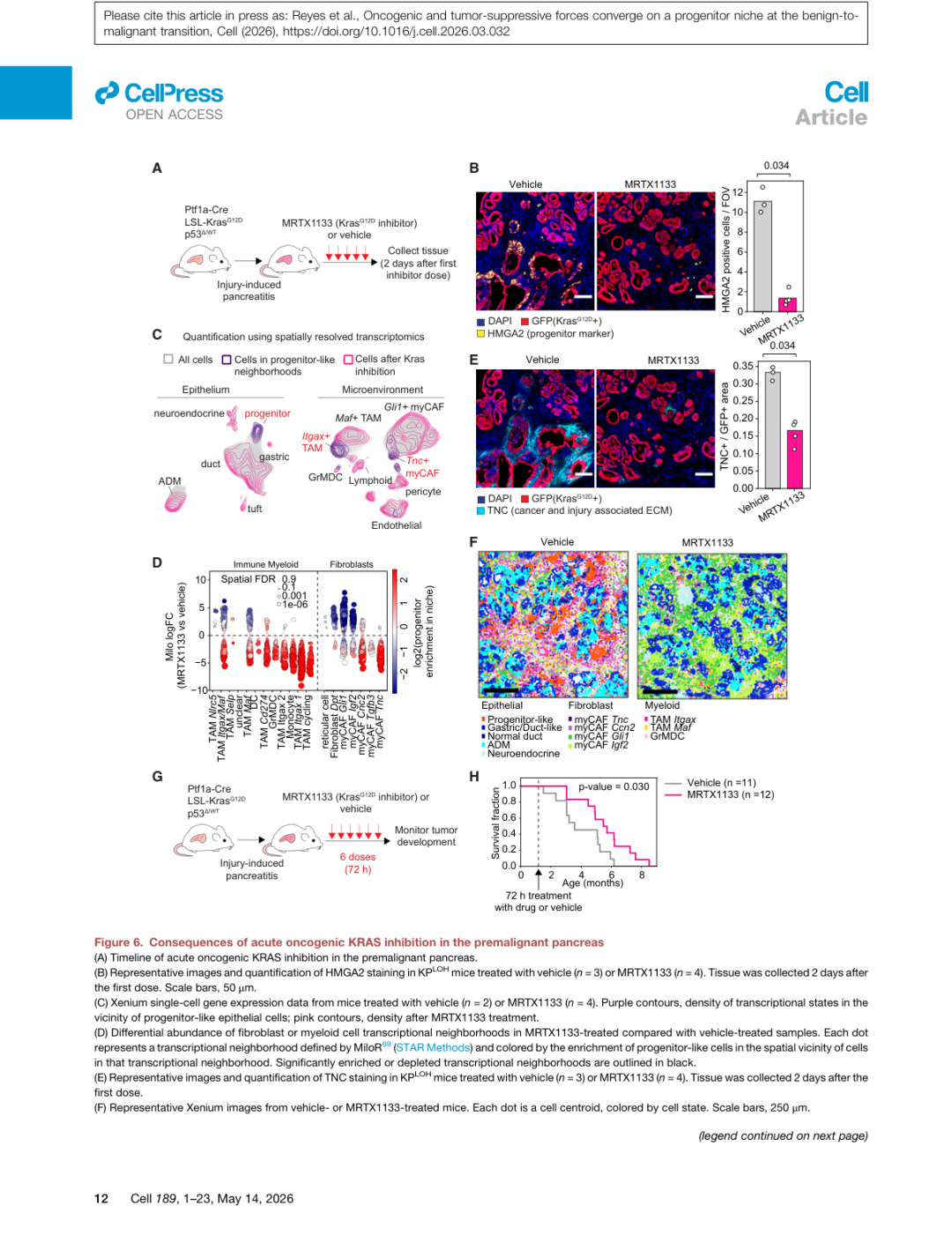
▲ 图6 | MRTX1133处理后,HMGA2+祖细胞样细胞数量急剧下降(B),TNC+肌成纤维细胞同步耗竭(E-F),Milo分析显示Tnc+/Itgax+ niche相关亚群选择性减少(D)。长期随访(G-H):仅72小时的KRAS抑制即可显著延缓PDAC发生(生存曲线,p=0.030),提示早期干预祖细胞niche的持久保护效应。
主要结果:
- MRTX1133处理后24小时内,44%的祖细胞样细胞出现凋亡,而胃样等其他亚群仅约6%凋亡——祖细胞样细胞对KRAS信号高度依赖
- scRNA-seq显示祖细胞样细胞减少24倍,显著高于其他亚群
- Tnc+肌成纤维细胞和Itgax+巨噬细胞同步崩解,niche在上皮祖细胞耗竭后迅速瓦解
- 长期随访:仅72小时的MRTX1133干预即可显著延缓PDAC发生(log-rank p=0.030)
八、p53:祖细胞清除的内源性执行者
p53在正常损伤修复中扮演什么角色?研究者利用KCshp53模型(可诱导的胰腺特异性p53短发夹RNA敲减)进行了反向实验。
结果显示,在胰腺炎后3周时:
- 对照组(shCtrl):祖细胞样细胞(HMGA2+)基本消退,组织恢复稳态
- p53敲减组(shp53):祖细胞样细胞持续存在,并向更间质化的"祖细胞2"状态演进(高表达Vim、YAP靶基因、IFN信号)
▲ 图7 | p53敲减后,祖细胞样细胞(HMGA2+)在胰腺炎后3周仍大量存在(B,p=0.0004),VIM+间质样细胞增多(E-F,p=0.013)。scRNA-seq揭示p53缺失驱动细胞沿G-P轴进一步漂移,形成"祖细胞2"亚态,伴随EMT、YAP、IFN-γ等致癌程序上调(C-D)。空间数据(H)显示,p53缺失导致祖细胞niche扩张为大片连续域,并伴随Itgax+/PD-L1high免疫抑制巨噬细胞大量积累——一个免疫豁免微环境形成。
通过整合multiome(scRNA + scATAC)数据,研究者精确刻画了p53在祖细胞中的直接靶向程序,包括:
- 细胞周期阻滞(Cdkn1a、Ccng1)
- 凋亡诱导(Bbc3、Bax、Perp)
- 衰老与DNA修复(Serpine1、Mgmt、Ercc5)
- 上皮稳态与修复(Itgb4、Sulf2、Areg)——这是p53在祖细胞中的非经典功能
p53并非直接抑制间质程序,而是通过清除高度可塑的祖细胞样状态,间接阻止了上皮-间质转化的发生。p53是"可塑性守门人",而非单纯的DNA损伤传感器。
p53缺失还带来微环境的深度重塑:Itgax+/PD-L1high(CD274high)巨噬细胞大量积累,形成免疫豁免niche;信号分析显示WNT(上皮→巨噬细胞)和SPP1(巨噬细胞→上皮)等信号轴在p53缺失后选择性激活,构成正反馈环路。
九、研究模型与临床启示
综合以上发现,本文提出了一个关于胰腺癌良恶性转化的整合模型:
Kras突变 + 组织损伤
↓
祖细胞样细胞出现(Msn/Hmga2/Nes+)
↓
同时触发:促癌程序(KRAS/EMT/糖酵解)
抑癌程序(p53/CDKN2A/SMAD4)
↓
┌──────────────┬──────────────┐
│ p53激活/KRAS抑制 │ p53失活 │
↓ ↓
祖细胞niche崩解 祖细胞niche扩张
组织稳态恢复 免疫豁免微环境形成
良性 良恶性转化→PDAC
临床转化意义:
- 早期干预窗口:祖细胞niche的形成早于可检测肿瘤,为癌症拦截提供了一个窗口期
- KRAS抑制剂的预防用途:短暂的KRASG12D抑制(如MRTX1133)即可产生持久的抗肿瘤效应,提示在高风险人群(如慢性胰腺炎、家族性PDAC)中预防性使用KRAS抑制剂的可能性
- 绕开p53修复的策略:由于直接恢复p53功能在技术上极为困难,靶向p53所天然消除的祖细胞样状态和癌样niche可能是更具操作性的治疗策略
- KRT17作为人类诊断标志物:KRT17+细胞在人类胰腺炎中的出现,及其与免疫抑制性微环境的相关性,为高风险病变的早期识别提供了潜在的生物标志物
- 巨噬细胞作为治疗靶点:anti-CSF1R可削弱祖细胞niche稳定性,可作为联合策略的一部分
十、小结
这项研究以前所未有的分辨率,在单细胞和空间层面解析了胰腺癌良恶性转化的核心机制。它的核心贡献在于:
- **定位了转化的"交锋现场"**:促癌与抑癌程序的对峙,不是弥散于整个癌前上皮,而是精确聚焦于一群罕见的祖细胞样细胞
- 揭示了niche的动态装配逻辑:祖细胞样细胞通过协调的多室细胞通讯,在肿瘤出现前就构建了癌样微环境
- 发现了p53的新功能维度:p53不仅维持基因组稳定性,更是上皮可塑性的守门人,通过清除祖细胞样状态来防止病理性niche的持久化
- 证明了预防性干预的可行性:短暂靶向KRAS即可瓦解祖细胞niche,并产生持久的防癌效应
良恶性转化,不是一蹴而就的突变事件,而是祖细胞样状态与其自我强化的多细胞niche协同演化、逃脱肿瘤抑制监控的生态系统演变过程。
这一框架不仅适用于胰腺癌,更为理解其他上皮性肿瘤的早期进展提供了普适性范式。
参考文献 Reyes J, Del Priore I, Chaikovsky AC, et al. Oncogenic and tumor-suppressive forces converge on a progenitor niche at the benign-to-malignant transition. Cell. 2026;189. https://doi.org/10.1016/j.cell.2026.03.032 IF: 42.5 Q1 B1
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-07,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号