Chimia | 基于深度学习的多肽设计

Chimia | 基于深度学习的多肽设计

DrugIntel
发布于 2026-05-08 19:22:14
发布于 2026-05-08 19:22:14
作者:Martin Pacesa(原载《Chimia》 2026年)
基于深度学习的多肽结合剂设计
摘要:多肽是由氨基酸组成的短链,天然介导着多种生物学功能,如细胞间信号传导、免疫调节、抗菌防御等。近年来,多肽作为一种极具吸引力的治疗模式崭露头角。它们兼具小分子药物和生物制剂的最佳特性,既能结合以往难以成药的分子表面,又保持了紧凑的结构并适合规模化生产。然而,设计多肽结合剂仍然充满挑战,因为它们的界面较小、暴露于溶剂中,且通常具有高度的动态性。深度学习的近期进展开始改变这一局面,使得针对特定靶标大规模生成并优先筛选多肽候选物成为可能。
1. 什么是多肽?为何它们如此引人关注?
首先,我所说的“多肽”(peptide)是指长度短于30个氨基酸的多肽,以此区别于稍大的“微型蛋白”(miniproteins,40–100个氨基酸)——目前的许多计算蛋白质设计流程主要是为后者开发的[1]。这个界限虽然有些武断,但我发现它很有用,因为在这个长度附近,我们开始观察到使多肽独具特色、且作为结合剂本质上更难设计的特性。这些分子在孤立状态下通常缺乏稳定的折叠构象,仅提供有限的相互作用表面,并且不再能像较大的蛋白质或抗体那样依赖同等程度的形状互补性和分布式结合能[2]。
多肽天然发挥着信号配体、激素、神经递质、抗菌效应因子、运输基序、毒素及识别元件的功能[3]。它们在生命之树上的广泛利用突显了其作为“低成本创新者”的进化价值:短而灵活的序列易于产生,可通过少量突变进行微调,能够快速扩散和周转,并通过构象可塑性作用于多种靶标[4]。从治疗角度来看,多肽恰好处于小分子和较大生物制剂之间的一个极具吸引力的中间地带。小分子主要靶向疏水口袋,但在广阔或无特征的蛋白质表面上举步维艰[5]。相比之下,抗体虽能以极佳的亲和力和特异性结合大多数靶标,但生产成本高昂,且主要局限于细胞外应用[6]。
多肽可以弥合这一鸿沟。它们为我们提供了更多的原子用于相互作用,大到足以接触延伸的蛋白质表面,同时又小到可以通过化学合成实现规模化制备。它们还可以通过环化、订书肽(stapling)、主链修饰、末端封端、聚乙二醇化(PEGylation)、脂化以及引入非经典氨基酸等手段进行调控[3]。此类修饰可显著提高其抗蛋白酶降解能力、构象预组织性、药代动力学性质、渗透性及选择性[3]。这表明无论是化学上还是结构上,多肽的空间都可以远远超越简单的线性氨基酸链。
2. 多肽结合剂的性质与挑战
从概念上讲,多肽结合剂的设计在根本上比大型蛋白质结合剂的设计更为困难,原因有几点(图1A)。首先是形式本身和界面尺寸。多肽可用于形成有利分子相互作用的表面积较少,因此每一个接触都十分重要,微小的建模误差都会带来更大的相对能量惩罚[2]。第二是熵。许多多肽在未结合状态下并非强预组织状态,这意味着有效的结合必须付出巨大的构象熵代价[7]。第三是溶剂暴露。由于多肽界面埋藏的表面积较小,溶剂相互作用与分子间接触的竞争更为激烈,这意味着每一个单独的相互作用对结合稳定性的贡献都更加关键。最后,多肽结合通常是动态的。重要的可能不是一种刚性结合的单一结构,而是一个部分预组织的构象集合、瞬态复合物以及诱导契合重排的集合[8]。目前的设计流程并未考虑到这一点,往往倾向于将问题简化为单个预测的复合物和少数几个置信度指标。
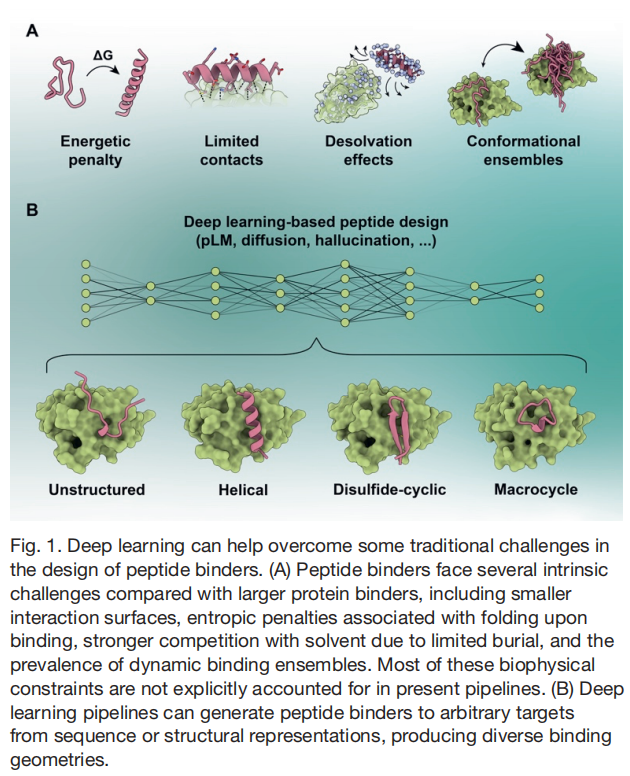
此外,多肽结合剂可以呈现不同的形式(图1B)。有些以两亲性螺旋的形式结合,有些则是β-发夹、伸展链或结构化环[2]。许多多肽自身可能基本无序,仅在结合时才变得有序[2]。一些多肽依赖于刚性化的环状或二硫键约束拓扑结构来降低熵成本[3],而另一些即使在结合状态下也可能至少部分保持“模糊”(fuzzy)状态[8]。这种多样性在多肽设计的讨论中常被低估。“多肽结合剂”并非单一的结构类别,一个在螺旋结合剂上表现良好的设计框架可能会在模糊相互作用剂或紧凑的大环肽上失效。这种异质性是该领域进展速度低于人们基于更广泛的蛋白质设计近期成功所预期的原因之一。
传统上,多肽结合剂是通过基于展示技术(display)或文库的筛选方法发现的,或是从天然来源中挖掘出来的。这些方法可能有效,但对结合模式的控制有限,且不易在初始命中后进行优化。更根本的是,当我们关心的属性(如亲和力和特异性)高度依赖于靶标、几何结构和系统时,纯粹的经验筛选是一种遍历巨大且化学多样性的空间的低效方式。计算设计为这一途径提供了更具吸引力的选择,因为它可以将搜索偏向于已经适应特定表面、结合模式或生化及药物属性的多肽。其真正的承诺不是取代实验,而是从一开始就让实验筛选变得更小规模、信息更充分、更具理性。
这正是深度学习改变格局的地方。此前,多肽设计受到制约,因为候选多肽必须被置于现实的结合几何构型中,然后以足够的准确度进行打分,才能从海量诱饵中区分出真正的结合剂[9]。深度学习通过学习来自蛋白质结构数据分布和序列共进化的多肽-蛋白质识别的可迁移约束改变了这一现状,这使得生成合理的设计方案变得更加容易。即便如此,可靠地识别真正的结合剂仍然是一个核心挑战,许多方法仍然依赖于不完美的预测和过滤预言机(oracles)[10]。
3. 已验证的多肽结合剂设计流程
一个新兴的方向主要依赖于蛋白质语言模型(pLMs),这些模型从大型序列数据库中学习蛋白质序列的潜在表征。这些模型直接从序列统计中捕捉与残基相容性、基序使用情况以及结构和相互作用倾向相关的模式[11]。因此,它们可以在不需要明确结构信息的情况下,基于靶标序列条件生成多肽候选物。诸如PepMLM[12]和PepPrCLIP[13]等方法通过生成基于靶标和习得共进化特征的多肽序列体现了这一方法。这很有吸引力,因为它不需要明确的靶标结构,因此可以应用于无结构或构象异质性的靶标。然而,仅凭序列进行设计仍然依赖于模型中隐含编码的相互作用特征,缺乏多肽-靶标相互作用环境的显式表示,这可能导致难以可靠地识别真正的结合剂。
第二类方法通过利用结构信息生成与目标界面兼容的多肽结合剂,更直接地解决了这一局限性。一个方向是使用基于扩散的生成模型,这些模型在三维空间中直接采样与目标结构条件相符的多肽主链。早期的例子是RFpeptides[14],它改进了RFdiffusion[15],生成几何上与目标界面兼容的大环多肽支架,随后进行序列设计。利用这种方法,研究人员获得了针对多个蛋白质靶标(包括MCL1、MDM2、GABARAP和RbtA)的多肽结合剂。这些大环肽表现出6 nM至2 μM的亲和力,且设计与晶体结构之间具有极高的结构一致性[14]。最近的框架将这一概念扩展到了更广泛的多肽结合剂模式。例如,BoltzGen在一个全原子框架中集成了结构预测和生成建模,能够设计多种类型的结合剂,包括线性多肽和二硫键环肽[16]。在实验活动中,针对RagC和RagA:RagC复合物获得了多肽结合剂,在测试数十种设计后产生了多个命中,其中最好的线性多肽表现出3.5 μM的亲和力,最好的大环肽约为80 μM。
一种应用更为广泛的多肽设计策略依赖于“幻觉设计”(hallucination)或预测器引导的优化。在此,结构预测网络被迭代用作设计预言机,以搜索满足预定义损失函数约束(如预测置信度、界面接触等)的多肽-靶标复合物序列-结构空间[17]。AfCycDesign代表了AlphaFold2[18]在环肽设计中的早期适配[19]。利用环状残差偏移,该流程可以有效模拟大环结构,用于设计针对MDM2和Keap1等靶标的结合剂,据报道抑制活性在纳摩尔至微摩尔范围内。相关方法如EvoBind[20–22]和EvoBind2[23]在AlphaFold2的引导下执行联合序列-结构搜索,并从多序列比对中提取共进化信息,能够仅利用靶标序列直接设计线性和环状多肽。EvoBind生成的环肽激动剂对GLP1R实现了32 nM的EC50[21],并对HIV包膜蛋白[20]和RNA酶A[23]表现出微摩尔级亲和力。同样,没有任何共进化先验的纯结构幻觉设计流程也被证明能够有效生成多肽结合剂。例如,BindCraft[24]被证明能够为致癌蛋白MDM2和治疗靶标WDR5生成具有纳摩尔级亲和力的螺旋多肽结合剂[25]。有趣的是,这些螺旋多肽可以通过化学订书(stapling)来锁定构象并提高结合亲和力。
最后,一个新兴的方向寻求将多肽设计扩展到经典氨基酸字母表之外。目前的大多数流程都在蛋白质结构预测模型使用的标准二十种残基范围内运行。RareFold[26]等新方法扩展了这一框架以纳入非经典氨基酸,使得设计含有扩展化学构建块的线性和环状多肽(微摩尔范围)成为可能,并允许探索传统蛋白质设计流程无法触及的相互作用化学。最近开发的几个生成框架(BoltzGen[16]、RFdiffusion3[27]、HalluDesign[28]、BoltzDesign[29]等)在全原子层面运行或允许任意化学约束,因此原则上能够纳入非经典残基,尽管这一能力尚未得到实验证实。
4. 填补空白
总的来说,我认为该领域正处于一个令人兴奋但仍处于发展阶段。毫无疑问,深度学习使得多肽结合剂设计比几年前更易实现且更具成效。我们现在已经有了可信的例子,证明计算方法能够生成经实验验证的线性和环状结合剂。然而,多肽的亲和力和命中率仍然低于我们对更大、结构更规整的结合剂的预期。这并不奇怪,因为目前的预测模型和下游过滤预言机在较小的界面、高度灵活的结合剂或非经典组分的多肽上表现仍然不佳[26,30]。在我看来,这现在是主要的瓶颈。
一个关键的局限性在于,大多数当前的流程并未显式建模多肽-蛋白质相互作用的物理决定因素。相反,它们依赖于源自结构预测模型的代理指标(proxy metrics),如预测置信度、界面接触或几何兼容性,来对候选复合物进行排序。虽然这些信号可以富集出合理的结合剂[31],但它们并未显式考虑结合的热力学或动力学,也未考虑结合态与非结合态之间的能量平衡。因此,溶剂竞争、与多肽折叠相关的熵惩罚以及构象异质性等重要贡献在很大程度上被忽略了。
因此,我怀疑下一阶段的进展不会仅仅来自现有流程的扩展或进一步优化,而是来自从根本上重新思考这些模型被训练来代表什么。改进对多肽化学和构象柔性的表征,可能是捕捉短多肽所能形成的多样化相互作用所必需的。这可能包括能够更好地表示多肽构象集合而非单一静态结构的模型,更显式地纳入溶剂和环境效应,并扩展到经典氨基酸字母表之外。沿着这些方向的进步可以让未来的设计框架更忠实地学习支配多肽-蛋白质识别的物理原理,并最终提高命中率和可达到的亲和力。
文献来源:M. Pacesa, Chimia 2026, 80, 265, DOI: 10.2533/chimia.2026.265.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号