Sci. Adv. | CryptoBank:面向隐秘结合位点的大规模数据库与序列预测模型

Sci. Adv. | CryptoBank:面向隐秘结合位点的大规模数据库与序列预测模型

DrugIntel
发布于 2026-04-28 13:05:27
发布于 2026-04-28 13:05:27

来源: Pedro Febrer Martinez et al., CryptoBank: A resource for the identification and prediction of cryptic sites in proteins.Sci. Adv.12,eady6364 (2026). 发表时间: 2026年4月22日 通讯作者: Francesco L. Gervasio(日内瓦大学 / 伦敦大学学院) 数据资源:www.cryptobankdb.com 代码链接:https://github.com/Gervasiolab/CryptoBank
一、研究背景与问题提出
1.1 "不可成药"蛋白的困境
传统药物发现高度依赖蛋白质表面明确的结合口袋——酶活性位点、离子通道孔道、已知变构位点等。然而,大量与疾病密切相关的蛋白质(如 RAS、MYC 等转录因子和信号蛋白)在其天然未结合(apo)状态下表面平坦、缺乏明显的空腔,被归类为"不可成药"靶点,极大限制了小分子药物的设计空间。
1.2 隐秘口袋(Cryptic Pocket)的概念
近年研究打破了这一认知范式。所谓隐秘口袋,是指在蛋白质自由态(apo 态)中不存在或不可探测,但在配体诱导或蛋白质自发构象波动后显现的结合空腔。其物理本质是:
- • 隐秘口袋对应蛋白质构象集合(conformational ensemble)中的高能态构象;
- • 这类构象在没有配体时出现概率极低,却可被小分子捕获并稳定;
- • 因此,隐秘口袋兼具直接抑制和变构调节两种药理学潜力。
1.3 现有方法的局限
此前用于发现隐秘口袋的方法主要分两类:
类别 | 代表方法 | 局限性 |
|---|---|---|
实验方法 | 高通量筛选(HTS)、片段筛选、NMR/X射线晶体学 | 成本高、通量低、大多属于偶然发现 |
计算方法 | 分子动力学(MD)模拟、增强采样(SWISH、FTMap)、ML模型(CryptoSite、PocketMiner) | 已有数据集规模极小(最大约1,500个结构),缺乏集合层面(ensemble-level)的隐秘性评估 |
核心瓶颈: 缺乏一个大规模、经过集合平均验证的隐秘口袋基准数据集,制约了预测模型的训练与评估。
二、CryptoBank 数据库的构建
2.1 数据收集与过滤流程
研究团队从 RCSB PDB(截至2025年8月11日)出发,实施了严格的多层筛选策略:
236,889 个 PDB 条目(X射线晶体学 + 冷冻电镜,分辨率 ≤ 2.5 Å)
↓ 过滤(含 UniProt 编号 + 95% 序列同一性簇归属)
157,327 个有效 PDB 条目
↓ 拆分为单链,分配至 apo / holo 集合
145,581 apo 链 + 168,916 holo 链
↓ 配对(同一95%同一性簇内)+ 纳入 AlphaFold 模型
81,302 apo 链 + 163,499 holo 链 → 19,781 个同一性簇
↓ 配体聚类(完全连接算法,7 Å 质心距离阈值)
34,014 个唯一配体 + 56,279 个唯一结合位点
↓ 结构比对(Cα RMSD ≤ 2.5 Å)+ 评分
6,000,000 个唯一 apo-holo-配体组合关键设计决策:
- • 将 AlphaFold 预测结构作为额外的 apo 链纳入,弥补了缺乏实验 apo 结构的蛋白质的覆盖空白;
- • 使用 Tanimoto 相似性为配体分配唯一标识符,确保相同化学实体跨链共享同一 ID;
- • 同一 holo 链中的每个配体单独处理,实现位点级别的精细分析。
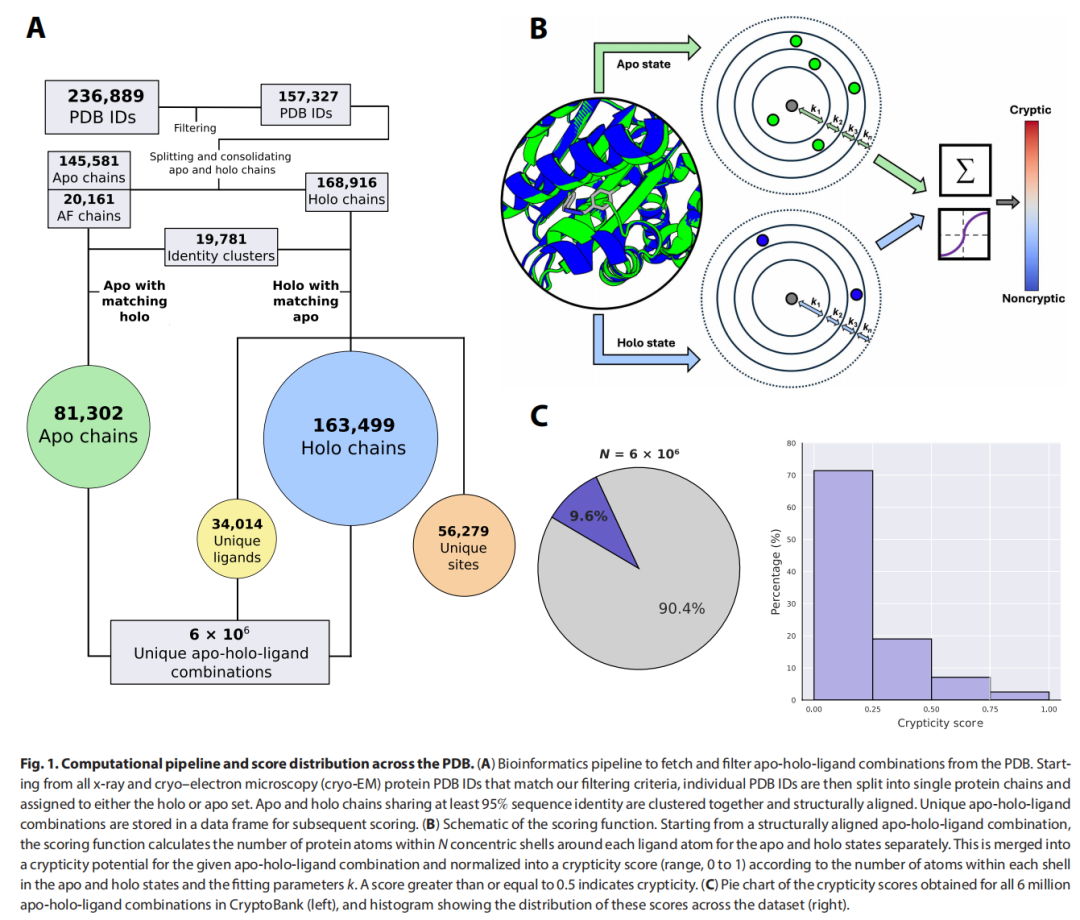
2.2 隐秘性评分函数(Crypticity Scoring Function)
评分函数的设计思想来源于一个物理直觉:若某结合位点是隐秘的,将配体叠加到 apo 结构时,配体原子将与 apo 态的残基产生严重的空间碰撞(clash);而在 holo 态中则几乎无碰撞。
具体地,对于每个 apo-holo-配体组合,模型执行以下计算:
第一步:壳层势能计算
将距离矩阵 (配体原子与蛋白原子间的成对距离,4 Å 截止)划分为 个同心球壳 ,每个壳层的势能为:
𝟙
其中 为可学习参数,𝟙 为指示函数。
第二步:系统能量聚合
第三步:配体分段与最终概率
为提升对结合区域不同部分的空间分辨率,将配体划分为 个片段,每段独立计算能量 ,最终以 sigmoid 函数聚合:
模型训练细节:
- • 训练集:199 个人工标注的 apo-holo 对(71 个隐秘,128 个非隐秘)
- • 正则化:L1 正则化抑制过拟合;5折交叉验证
- • 超参数搜索:(配体分段数),(壳层数),(正则化强度)
- • 最终准确率:89%(独立测试集)
- • 判定阈值:评分 ≥ 0.5 即判定为隐秘构象
2.3 集合层面的位点隐秘性评分
为克服单一 apo 结构的局限性(不同晶体学条件可产生不同的 apo 构象),研究团队对同一结合位点的所有 apo-holo-配体组合取平均评分,作为位点级别的集合隐秘性评分(site crypticity score)。
此外,采用异常值导向策略(outlier-oriented strategy):对均值评分低于0.5、但存在少量高隐秘性异常构象的位点进行回收,避免遗漏"稀有但真实"的隐秘位点。
三、数据库统计特征与药化学分析
3.1 总体规模
指标 | 数值 |
|---|---|
分析的 apo-holo-配体组合总数 | ~6,000,000 |
判定为隐秘构象的组合数(评分 ≥ 0.5) | ~574,000(9.6%) |
隐秘结合位点总数 | 5,151(占全部位点的 9.2%) |
涉及的95%同一性蛋白质簇数 | 3,643(占全部簇的 18.4%) |
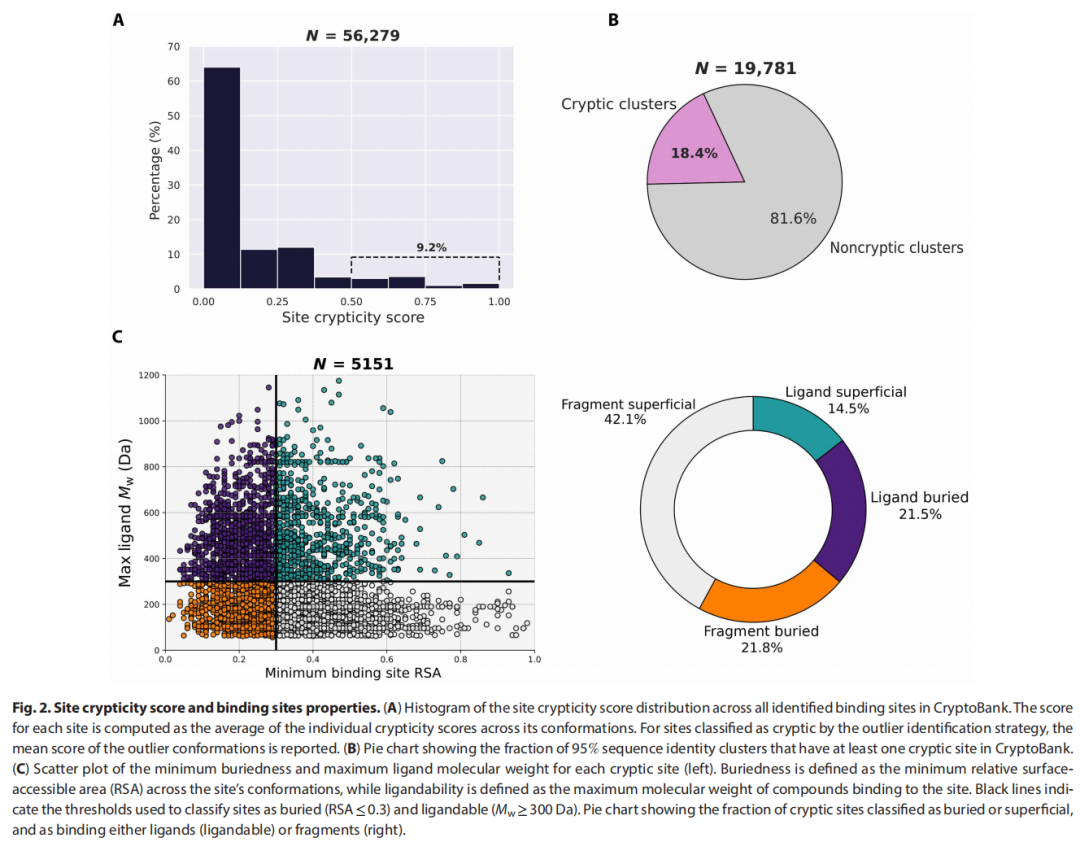
3.2 配体分子量与埋藏程度分类
研究根据两个维度对隐秘位点进行分类:
- • 配体大小:片段类( Da)vs 配体类( Da)
- • 位点埋藏深度:埋藏型(相对溶剂可及面积 RSA < 0.3)vs 表浅型(RSA > 0.3)
类别 | 比例 | 药学意义 |
|---|---|---|
片段-埋藏型 | 21.8% | 需要蛋白质重排才能进入,最具成药价值 |
片段-表浅型 | 42.1% | 多个化学多样片段结合区域提示隐秘性 |
配体-埋藏型 | 21.5% | 直接成药价值高(60% 的配体结合位点为埋藏型) |
配体-表浅型 | 14.5% | 部分可作为变构调节靶点 |
值得注意: 约 77% 的配体类隐秘位点所结合配体分子量在300–500 Da,符合 Lipinski 类药五原则(rule-of-five),提示这些位点具有直接的临床转化潜力。
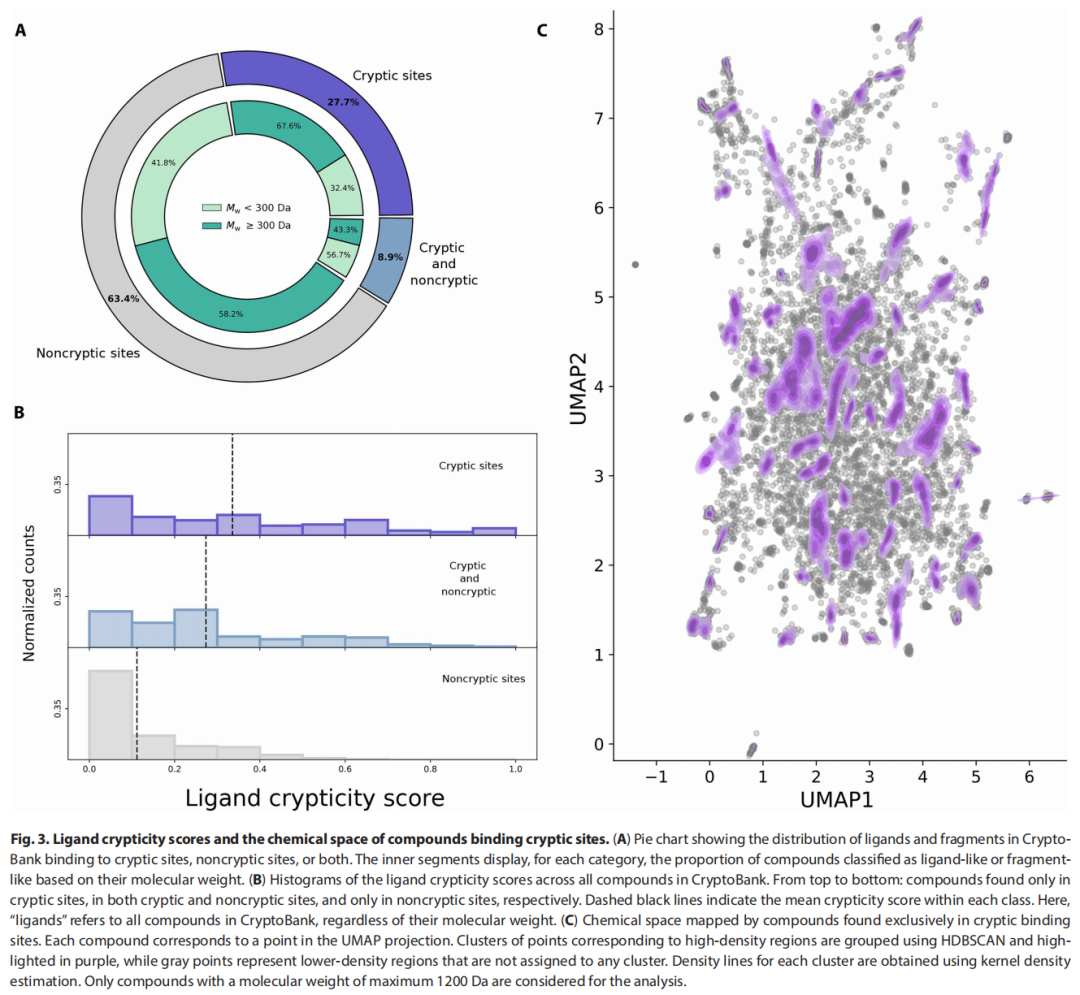
3.3 配体化学空间分析与片段库设计
研究团队对9,400个仅出现在隐秘位点的配体进行了系统的化学空间分析:
- • 利用 Morgan 指纹相似性 + UMAP 降维 + HDBSCAN 聚类,识别出 60个结构多样性簇
- • 簇间最大 Tanimoto 相似性仅为 0.35,表明化学多样性极高
- • 这60个代表性分子可作为针对隐秘口袋的专属片段库的设计起点
此外,利用"相似蛋白倾向结合相似配体"的原则,CryptoBank 支持靶标特异性片段库的定制化生成,并可以配体隐秘性评分对筛选候选分子进行排序。
四、与药学相关靶点的交叉分析
4.1 与 Open Targets 平台的交叉
将 CryptoBank 与 Open Targets 数据库(疾病关联评分 > 0.5)交叉后:
- • 约6,000个疾病相关基因中,有 1,567 个独立簇在 CryptoBank 中有对应
- • 其中 33.8% 的疾病相关蛋白含有隐秘位点
- • 在已有上市药物或在研药物(仅考虑小分子/PROTAC类)的133个单结合位点靶蛋白中,26.3% 的结合位点具有隐秘性
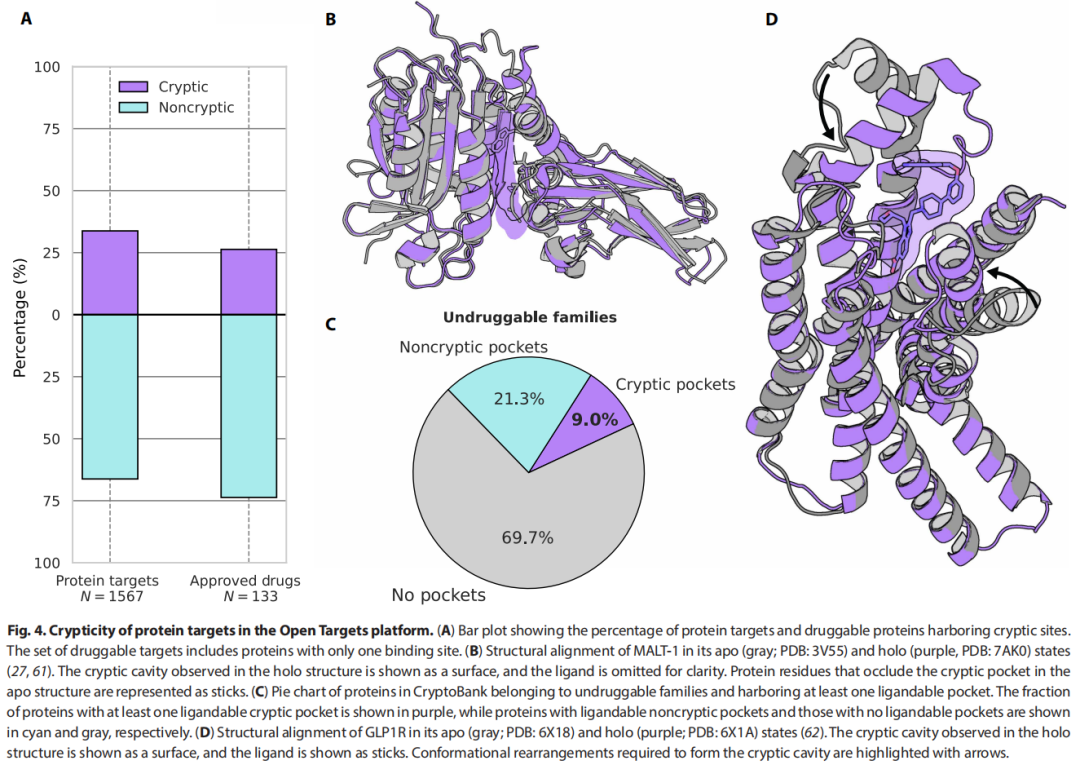
4.2 典型案例
MALT-1(黏膜相关淋巴组织淋巴瘤转位蛋白1)
- • CryptoBank 识别出其 caspase 结构域中的一个变构隐秘口袋
- • 该位点当前正处于 1期临床试验(NCT05544019),用于治疗复发/难治性 B 细胞肿瘤
- • 验证了 CryptoBank 捕捉具有临床意义的隐秘位点的能力
GLP1R(胰高血糖素样肽-1受体)
- • apo/holo 结构比对清晰显示:配体结合诱导α螺旋部分解折、环区位移及另一α螺旋的内移,形成隐秘口袋
- • 与当前主流 GLP-1 类药物(如司美格鲁肽)的靶点高度相关,提供新的变构调节视角
4.3 "不可成药"蛋白的重新评估
在 Open Targets 数据库标记为"不可成药家族"的蛋白中(筛选条件:RSA < 0.3 且结合25–50个重原子的配体):
- • 30.3% 含有埋藏且可配体化的结合位点
- • 其中 9% 的蛋白含有潜在可成药的隐秘口袋
这一结果直接支持了"隐秘口袋可系统性扩展可成药蛋白质组"的核心论点。
五、蛋白质语言模型(PLM)微调与隐秘位点预测
5.1 模型选择与微调策略
研究团队选用 ProtTrans Prot-T5-XL-UniRef50 作为基础模型:
- • 预训练于4,500万条蛋白质序列(140亿氨基酸)
- • 采用**低秩自适应(LoRA)**向注意力机制注入可训练参数,实现参数高效微调
- • 在输出端添加卷积神经网络预测头(1D卷积层,512个输出通道,核尺寸3 → 线性分类层)
- • 任务定义:残基级二分类(每个残基是否属于隐秘结合位点)
5.2 训练数据集构建
数据集切分策略(防止序列同一性泄漏):
- • 使用 MMseqs2 以20%序列同一性进行聚类(
--min-seq-id 0.2, --cluster-mode 3) - • 按连通分量(connected component)整体分配至同一数据集,确保测试集中任意序列与训练/验证集的相似性 < 20%
数据集 | 序列数量 | 说明 |
|---|---|---|
训练集 | 6,345 | 来自3,643个隐秘簇 |
验证集 | 793 | 与训练集 <20% 同一性 |
测试集 | 793 | 与训练/验证集 <20% 同一性 |
- • 正类(隐秘残基)占比仅约 5%,属于高度不平衡分类任务
5.3 模型性能评估
指标 | 训练集 | 验证集 | 测试集(<20% 同一性) |
|---|---|---|---|
ROC AUC | 0.97 | 0.92 | 0.70 |
PR AUC | 0.79 | 0.65 | 0.11 |
相对随机预测提升 | >10× | 数量级 | >2× |
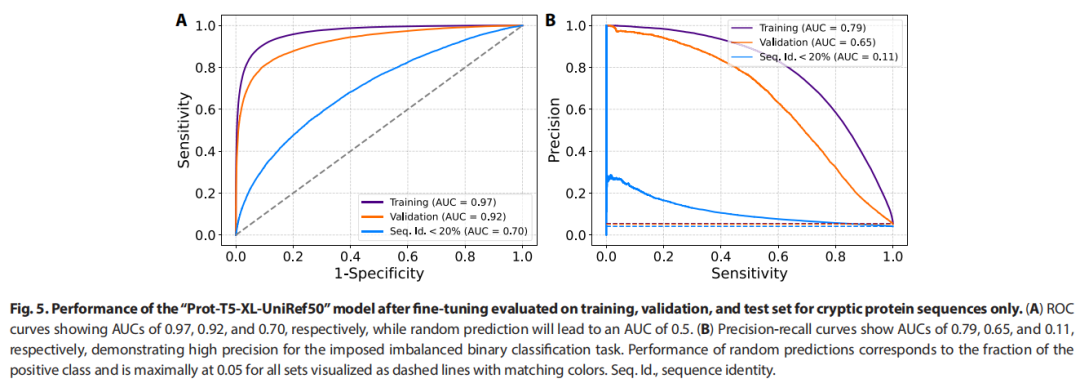
关于 PR AUC 的说明: 由于正类比例极低(约5%),ROC AUC 在不平衡场景下容易高估模型性能,而 PR AUC 是更严格、更真实的性能衡量指标。测试集 PR AUC 为0.11,较随机预测(~0.05)提升超过两倍,表明模型对未知序列仍有一定泛化能力。
适用性边界总结:
查询序列与 CryptoBank 同一性 > 20% → ROC AUC ≈ 0.92,PR AUC ≈ 0.65(高置信预测)
查询序列与 CryptoBank 同一性 < 20% → ROC AUC ≈ 0.70,PR AUC ≈ 0.11(初步筛查)六、PLM 预测与分子动力学模拟验证
研究团队选取四个不在训练集中的蛋白质进行预测验证,所用 MD 框架:GROMACS 2023 + PLUMED 2.9,分子力场 DES-Amber 或 AMBER99-ILDN,水模型 TIP4P-D 或 TIP3P;增强采样采用 SWISH-X 方法(6个并行副本,含1M苯作为余溶剂,OPES 多温度组件,温度范围300–350 K)。
6.1 HyBcl-2-4(水螅 Bcl-2 同源蛋白)
- • 与人类 Bcl-XL 序列同一性 37.5%,整体折叠保守(RMSD < 2 Å)
- • PLM 预测在 HyBcl-2-4 与 HyBak1 BH3 的结合界面存在隐秘口袋
- • 人类 Bcl-XL 在该界面的隐秘口袋已有晶体学验证;HyBcl-2-4 此前无相关报道
- • SWISH-X 证实口袋开放;无偏 MD 未能发现,与人类同源蛋白的已知行为一致
6.2 TPP1(端粒蛋白)
- • 参与端粒酶招募与激活,是重要的治疗靶点
- • PLM 预测在远离蛋白-蛋白相互作用界面的区域(α螺旋及其连接环)存在变构隐秘位点
- • 冷冻电镜结构(PDB: 7TRE)显示蛋白核心内部存在深埋空腔,但如何变为溶剂可及不明
- • 无偏 MD 中预测区域形成溶剂暴露腔;SWISH-X 进一步证明该区域的α螺旋倾斜和连接环外移可将深埋空腔与蛋白表面相连通,形成完整的通路
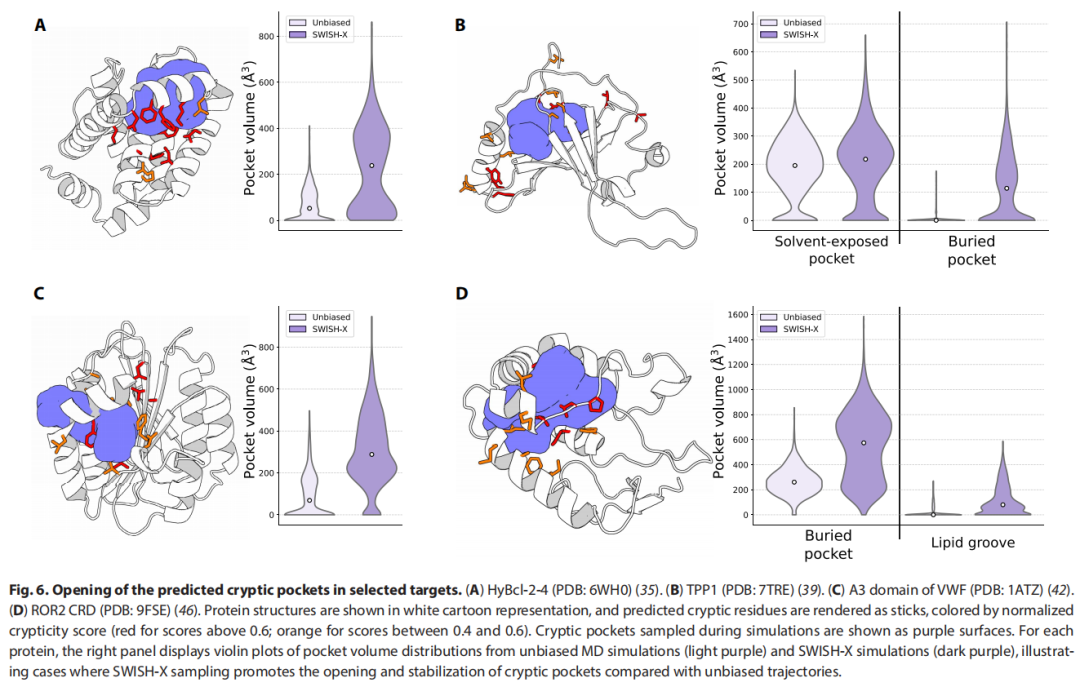
6.3 VWF A3 结构域(血管性血友病因子 A3 结构域)
- • 介导血小板黏附(通过与胶原I/III结合)
- • PLM 识别出7个高隐秘性评分残基(>0.6),位于胶原结合面的对侧,夹持在两个α螺旋与β折叠之间
- • 4个已报道的高分辨率晶体结构中该区域均无明显口袋
- • 一个抗体结合结构(PDB 中含乙酸分子的结构)在预测区域内存在微小空腔——与预测吻合
- • SWISH-X 模拟显示 C 端6残基的α螺旋发生位移,口袋体积显著增大
6.4 ROR2 CRD(受体酪氨酸激酶 ROR2 的半胱氨酸富集结构域)
- • PLM 预测15个残基(归一化评分 > 0.6)分布于3个α螺旋及1个连接环
- • 该区域在其他受体家族(Frizzled 类、NRK CRD)中介导脂质/辅因子结合,但 ROR2 的晶体结构(PDB: 9FSE)未报告类似特征
- • SWISH-X 模拟揭示:三螺旋间可形成沟槽样特征,空间足以容纳脂质配体;提示一种与 Frizzled 类受体类似的配体识别机制
七、局限性与未来展望
7.1 当前局限
局限 | 说明 |
|---|---|
PDB 数据偏差 | 数据来源依赖实验结构,可能偏向特定蛋白家族或结晶条件,影响代表性 |
PLM 泛化能力有限 | 对 <20% 同一性序列的预测精度显著下降(PR AUC 降至0.11),高度新颖蛋白的预测仍具挑战 |
缺乏序列外结构信息 | PLM 仅利用序列信息,未整合三维结构特征,可能错过结构层面的关键信号 |
隐秘性定义的构象依赖性 | 所有评分均基于已有实验结构的 apo/holo 对,对于实验结构稀缺的蛋白质覆盖不足 |
7.2 未来方向
- • 将结构信息整合进预测模型,突破序列相似性壁垒
- • 扩展至冷冻电子断层扫描等新兴结构数据源,提升构象多样性覆盖
- • 与实验片段筛选形成闭环,持续丰富数据库
- • 开发针对隐秘口袋的从头配体设计(de novo design)工作流程
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号