Nat. Chem. Eng. | 端到端反应性预测框架:重塑多相催化

Nat. Chem. Eng. | 端到端反应性预测框架:重塑多相催化

DrugAI
发布于 2026-03-30 18:47:56
发布于 2026-03-30 18:47:56
DRUGONE
多相催化反应通常由复杂的多步反应网络组成,不同反应路径之间存在竞争关系,因此理解完整反应机理对于预测催化活性和选择性至关重要。传统方法依赖研究人员基于经验提出反应步骤,并使用密度泛函理论计算反应能量,但这种方式难以覆盖所有可能路径,尤其是在反应网络规模迅速增长时。
研究人员提出一种用于多相催化反应性的端到端计算框架 CARE(Catalytic Automated Reaction Evaluator),该框架结合规则驱动的反应网络生成、基于机器学习的热力学与动力学参数预测以及快速微观动力学求解,实现从反应路径生成到反应速率预测的完整自动化流程。该方法能够再现实验趋势,并成功模拟甲醇分解、CO₂电还原以及费托合成等复杂反应体系,证明端到端建模可以探索传统原子级模拟难以处理的大规模反应网络。

多相催化过程依赖于一系列相互竞争的基本反应步骤,这些步骤共同构成复杂的化学反应网络。每个反应网络由反应物、中间体和反应路径组成,其反应速率和选择性取决于各个步骤的能量和动力学参数。
传统机理研究通常依赖人工提出反应路径,再通过密度泛函理论逐步计算能量。这种方法在简单体系中有效,但随着反应网络规模增加,可行路径数量呈指数增长,使得人工构建机理变得困难。此外,传统计算往往只研究最稳定表面,而真实催化剂通常包含多种晶面,因此预测结果与实验存在差异。
为了解决这一问题,研究人员发展了自动化反应网络算法。这类方法可以自动生成反应路径,但通常需要大量第一性原理计算,计算成本极高,限制了其在复杂催化体系中的应用。近年来,机器学习被用于加速能量预测,但现有方法往往只能处理局部反应或少量中间体,难以与完整反应网络结合。
因此,需要一种能够自动生成反应网络、快速预测反应能量并直接计算反应速率的统一框架,从而实现真正的端到端催化建模。
方法
研究人员提出的CARE框架由三个核心模块组成。
第一部分是基于规则的反应网络生成器。该模块根据预定义的化学反应模板自动生成所有可能的中间体和反应路径,从而构建完整的反应网络。与传统依赖第一性原理逐步探索的方法不同,该策略可以在不进行昂贵计算的情况下快速生成大规模反应空间。
第二部分是热力学和动力学参数预测模块。研究人员利用先进的机器学习模型对反应能量、吸附能以及过渡态能垒进行预测,从而替代大规模密度泛函理论计算。该模块能够在保证精度的同时显著降低计算成本。
第三部分是微观动力学求解器。该模块根据预测得到的反应参数求解完整反应网络的动力学行为,得到反应速率、选择性以及不同条件下的催化性能。
通过将反应生成、能量预测和动力学模拟整合在同一框架中,CARE实现了真正的端到端催化反应建模。
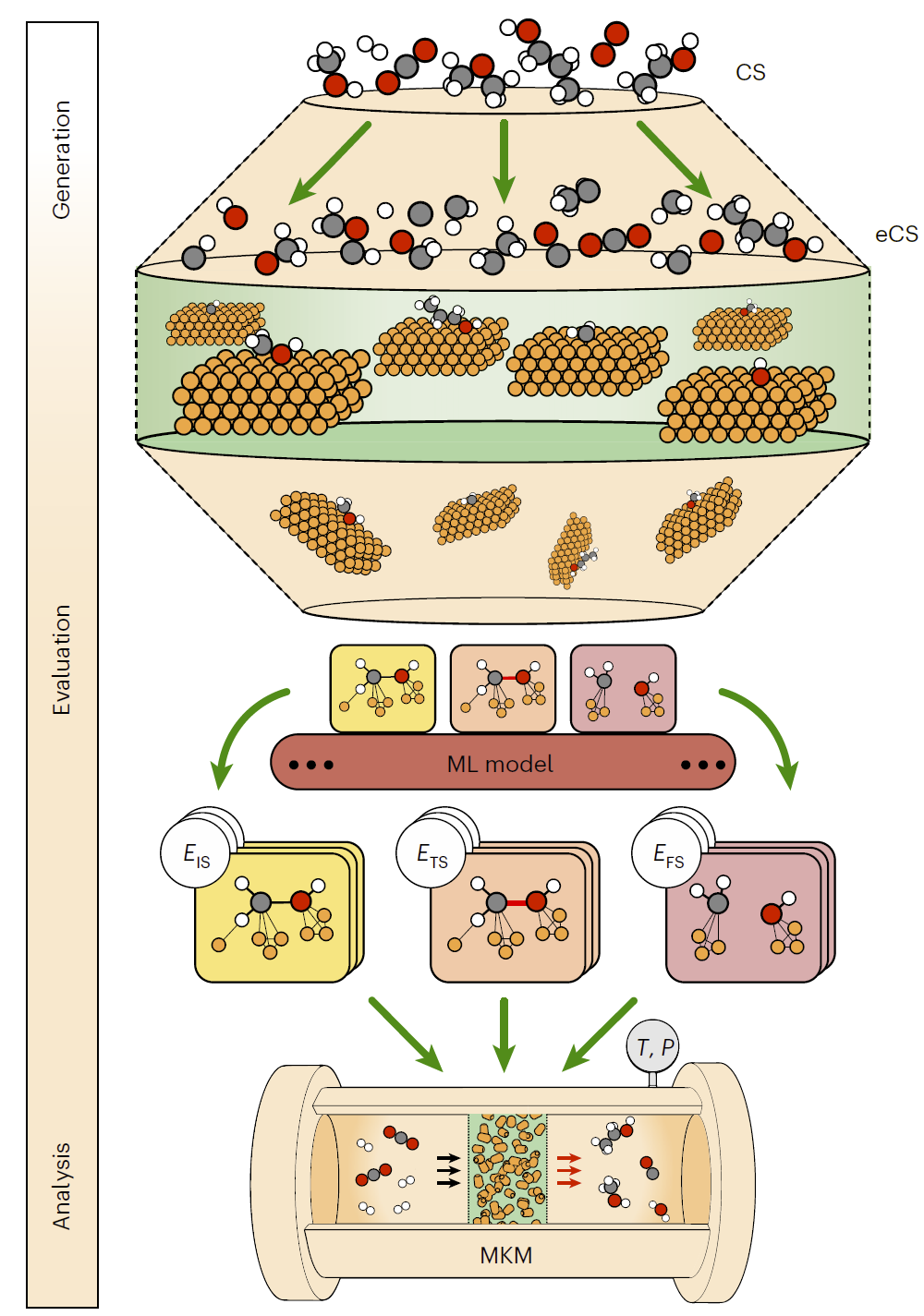
图1: CARE流程示意:从反应网络生成到反应性分析的完整工作流程。
结果
反应能量预测精度
研究人员首先验证机器学习模型在反应能量预测中的精度。结果表明,该模型在吸附态和过渡态能量预测中误差较小,能够满足催化反应建模的要求,并显著降低计算成本。
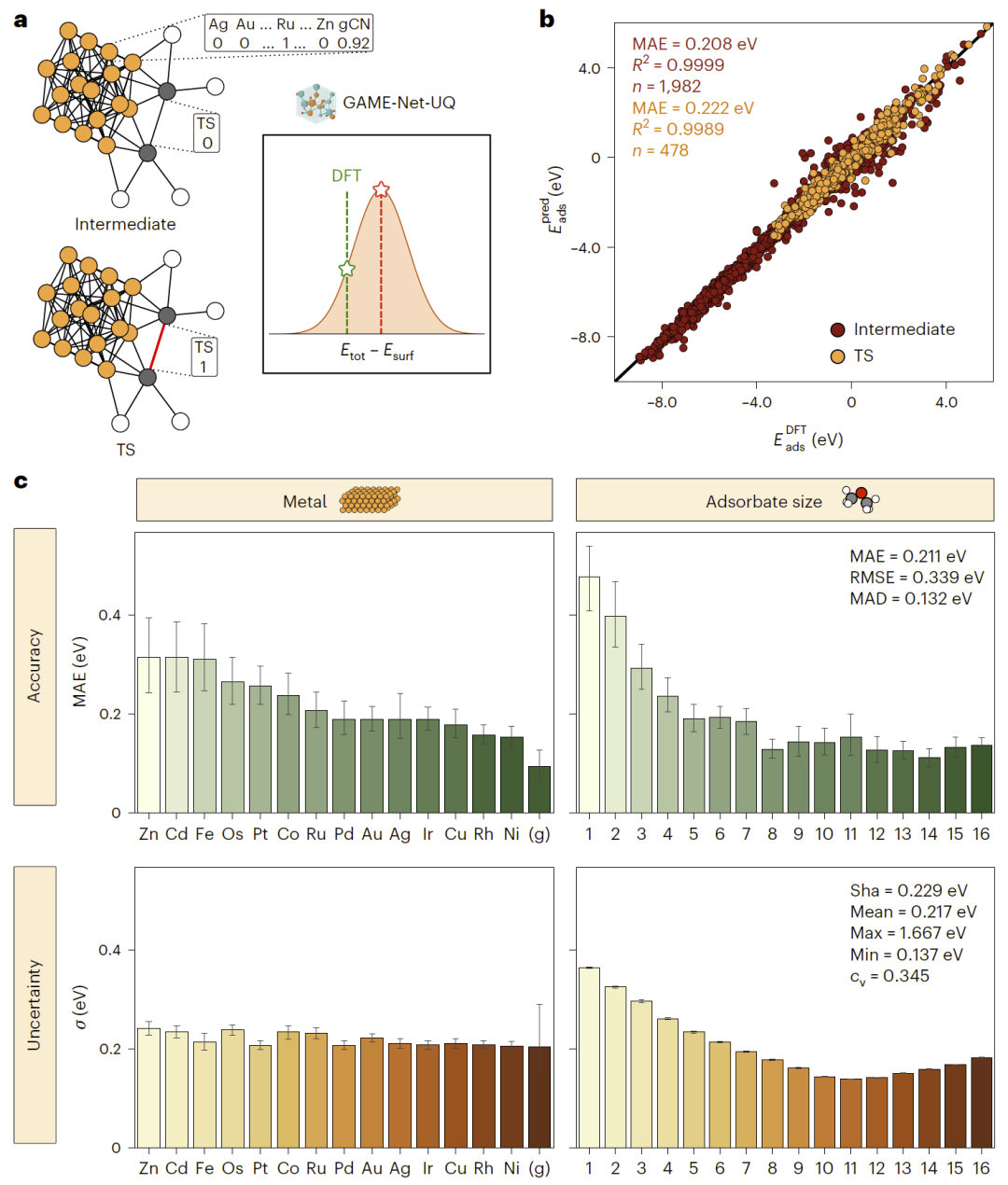
图2: GAME-Net-UQ模型性能评估结果。
多表面催化活性预测
在金属表面催化反应测试中,CARE能够同时考虑多个晶面,并预测整体催化活性。计算结果再现了实验中不同金属催化活性的趋势,说明端到端框架能够真实反映催化剂性能。
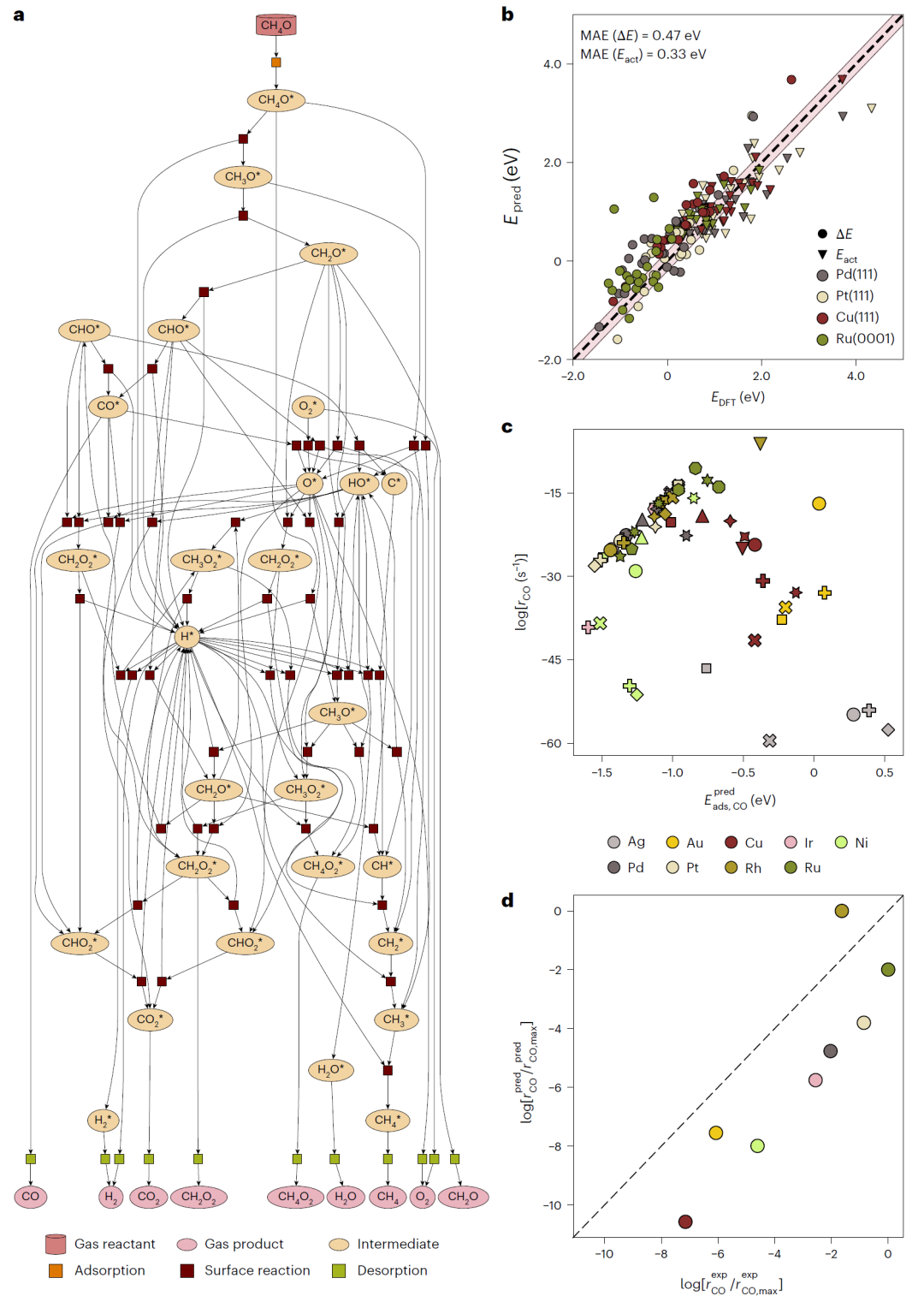
图3: 甲醇分解反应机理与催化活性分析。
CO₂电还原反应网络
研究人员进一步使用CARE模拟CO₂电还原反应,构建包含大量中间体的反应网络,并预测不同条件下生成C₃产物的选择性。结果显示,该框架能够处理电催化体系,并捕捉反应条件对选择性的影响。
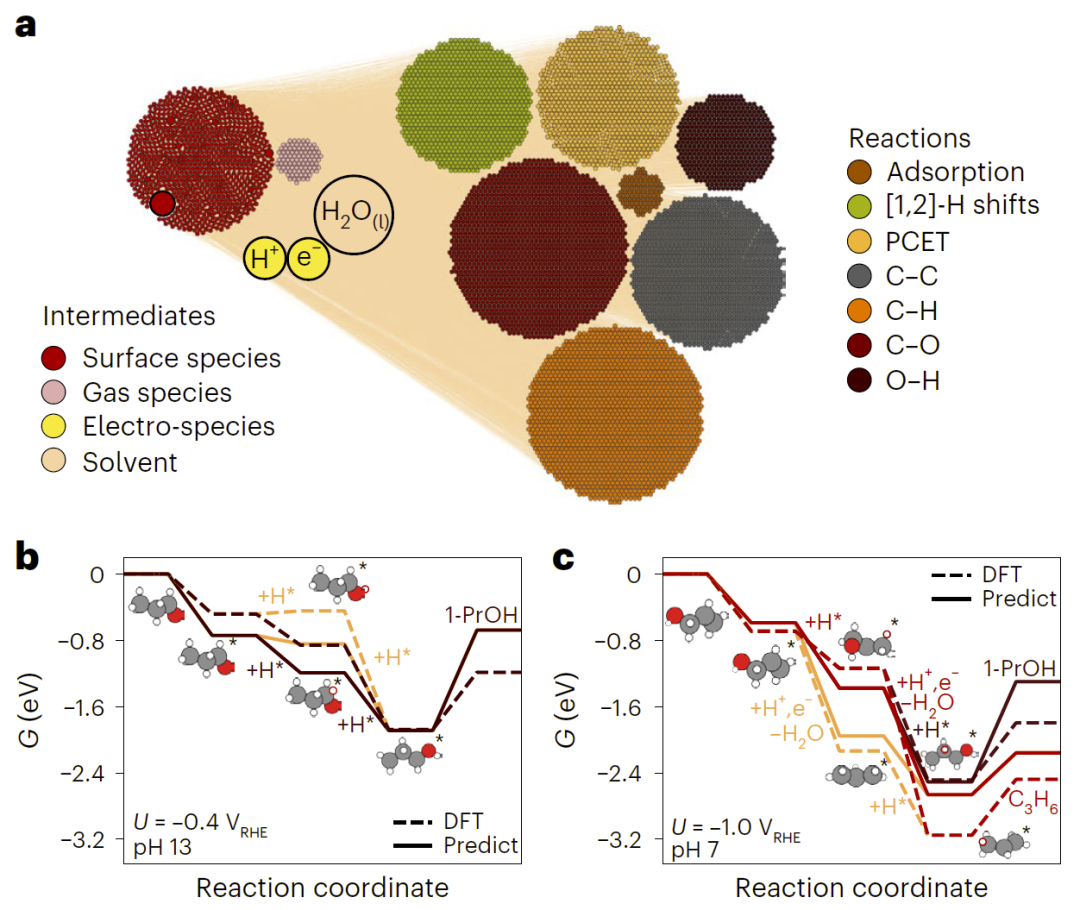
图4: Cu(100)表面上电化学 C₃O₂ 反应网络。
费托合成大规模反应网络
在费托合成模拟中,CARE生成包含数十万条反应路径的反应网络,并成功预测生成C₆产物的机理。这一规模远超传统第一性原理计算所能处理的范围,展示了端到端框架在复杂体系中的优势。
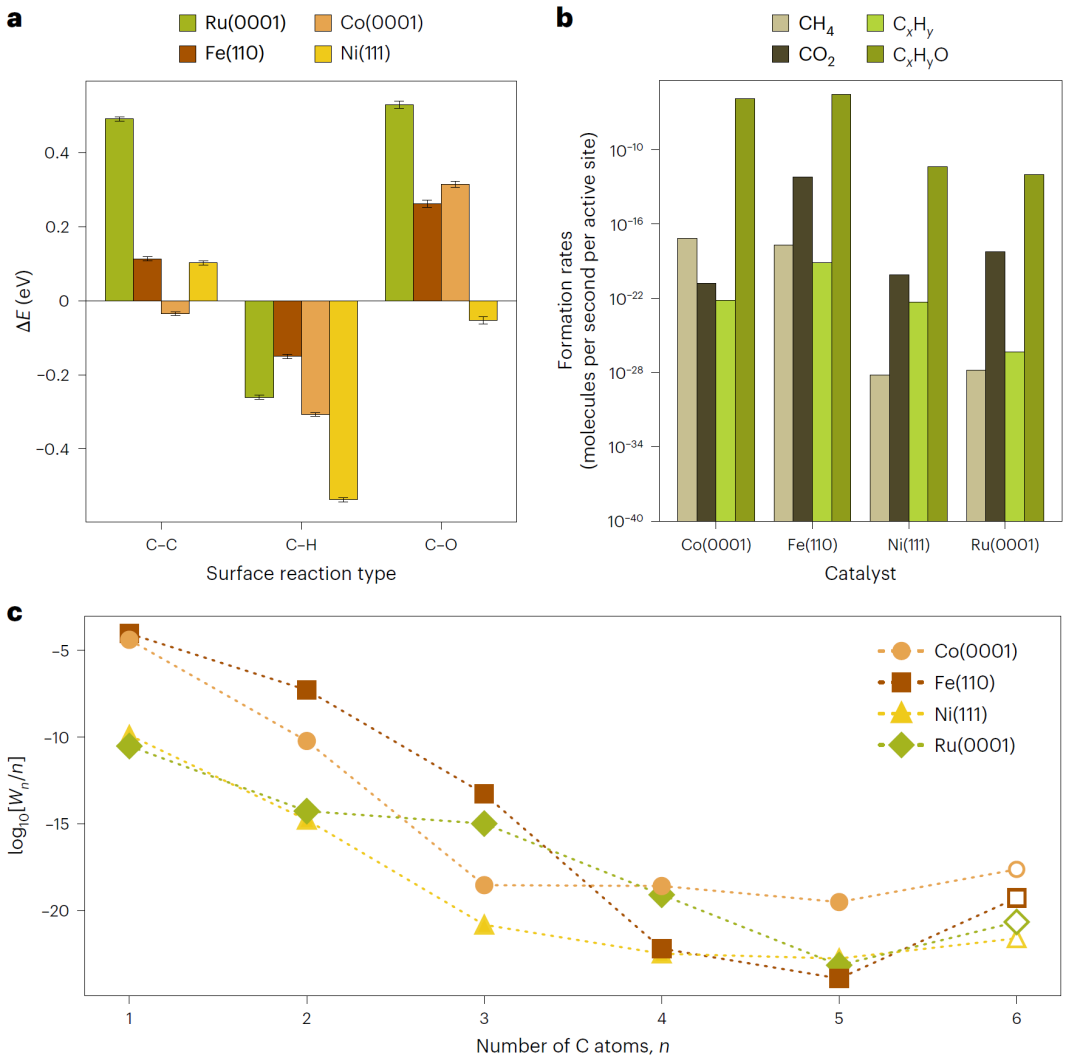
图5: 费托合成 C₆O₁ 反应网络得到的能量与动力学趋势。
讨论
本研究提出的CARE框架实现了多相催化反应从反应路径生成到反应速率预测的全流程自动化。通过结合规则驱动的反应网络生成、机器学习能量预测以及微观动力学模拟,研究人员能够在大规模反应空间中快速筛选催化机理。
结果表明,该框架不仅能够再现实验趋势,还可以探索传统方法难以研究的复杂反应体系,包括多晶面催化、电催化以及长链产物生成反应。
研究人员认为,端到端建模是未来催化计算的重要方向。随着机器学习模型和计算资源的提升,该方法有望实现对真实工业催化体系的预测,并加速新催化剂的设计。
未来工作将进一步提高能量预测精度,并扩展到更复杂的反应体系,从而实现真正的高通量催化机理探索。
整理 | DrugOne团队
参考资料
Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng (2026).
https://doi.org/10.1038/s44286-026-00361-8

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-20,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号