Nature | 超大规模虚拟筛选发现新化学骨架:一项重塑药物发现边界的里程碑研究

Nature | 超大规模虚拟筛选发现新化学骨架:一项重塑药物发现边界的里程碑研究

DrugIntel
发布于 2026-03-30 16:00:08
发布于 2026-03-30 16:00:08

原文:Lyu J, Wang S, Balius TE, Singh I, et al. Ultra-large library docking for discovering new chemotypes. Nature. 2019;566:224–229. DOI:10.1038/s41586-019-0917-9 通讯作者:Brian K. Shoichet(UCSF)、Bryan L. Roth(UNC Chapel Hill)、John J. Irwin(UCSF) 发表时间:2019年
目录
- 1. 研究背景与核心问题
- 2. 技术框架:从虚拟库构建到大规模对接
- 3. 靶标一:AmpC β-内酰胺酶筛选
- 4. 靶标二:D4 多巴胺受体筛选
- 5. 命中率-打分曲线:虚拟筛选预测能力的量化验证
- 6. 人工评估 vs 自动化筛选
- 7. 关键方法论细节
- 8. 研究局限性
- 9. 总结评述
一、研究背景与核心问题
1.1 类药化学空间的规模与悖论
1996年,Bohacek 等人在一篇被广泛引用的文章中估算,类药分子(drug-like molecules,MW < 500 Da,类药五规则范围内)的数量超过 10⁶³。相比之下,有史以来合成并收录于数据库的化合物总量约为 10⁸ 量级(如 PubChem 1.1亿,ZINC 7.5亿),二者之间存在数十个数量级的鸿沟。
这一悖论的实践含义是:现有药物发现所探索的化学空间,相对于理论可行空间而言几乎是零。 早期药物发现中,初始命中化合物的结构特征往往直接决定后续优化的方向和天花板("hit-to-lead"依赖性),因此扩展前期筛选的化学多样性具有战略意义。
1.2 现有解决方案的局限
在本文发表之前,扩展筛选规模的主要策略包括:
策略 | 代表性方案 | 主要局限 |
|---|---|---|
实体高通量筛选(HTS) | 企业内部化合物库(10⁵–10⁶量级) | 规模受储存/成本限制;多样性相对有限 |
DNA编码化合物库(DEL) | GSK、X-Chem 等 | 依赖特定反应类型,骨架多样性受限;难以验证结合模式 |
传统虚拟筛选 | 商业化合物库对接(ZINC in-stock,~350万) | 库规模小;受限于现货化合物 |
枚举式组合库 | 侧链组合扩展 | 骨架多样性不足;合成验证困难 |
本文的核心创新 在于:将"按需合成"(make-on-demand)理念与超大规模结构对接相结合,突破了"只能筛选现货化合物"的传统瓶颈。
1.3 研究假说与核心问题
论文围绕三个递进的科学问题展开:
- 1. 随着虚拟库规模从百万扩展至亿级,对接的富集能力(enrichment)是否会因噪声淹没信号而退化?
- 2. 在超大规模库中,能否发现对于给定靶点而言在小库中不存在的全新化学骨架?
- 3. 对接打分(docking score)是否能定量预测实验命中率,从而建立可用于预测活性化合物总量的数学模型?
二、技术框架:从虚拟库构建到大规模对接
2.1 按需合成库(Enamine REAL 库)的构建原理
本研究使用的虚拟库来源于 Enamine 的 REAL(Readily Accessible)化合物库,其构建逻辑如下:
- • 构建单元(Building blocks):约 7 万种经过质量认证的现货砌块分子
- • 反应类型:130 种经过充分验证的双组分反应(酰胺偶联、Suzuki 偶联、还原胺化、磺酰化等主流反应)
- • 枚举策略:对所有合法砌块组合进行穷举,仅保留满足类先导物(lead-like)或类药性标准的产物
论文发表时,该库包含约 1.7 亿个分子(AmpC 筛选时为 9900 万,D4 受体筛选时扩充至 1.38 亿);截至 2019 年底,已超过 3.5 亿;预计 2020 年突破 10 亿。每新增约 20 个化合物,就引入一个全新 Bemis-Murcko 骨架,最终形成超过 1070 万种独特骨架。
关键特征:库内化合物高度"功能性拥挤"(functionally congested),具有较强的三维性(3D character),与传统扁平芳香类化合物库形成鲜明对比;少于 3% 的化合物可从其他来源商购获得,确保了真正的新颖性。
2.2 对接前处理流程
对接前的分子准备流程包括:
- 1. 质子化状态与互变异构体:使用 JChem(v15.11.23.0)在实验 pH(接近中性)条件下计算,生成最可能的质子化形式
- 2. 三维构象生成:使用 Corina(v3.6.0026)生成初始三维坐标
- 3. 构象集合枚举:使用 OMEGA(v2.5.1.4)生成低能构象集合
- 4. 原子电荷与去溶剂化能:使用 AMSOL 计算 AM1-BCC 部分电荷,并计算配体去溶剂化惩罚项
2.3 DOCK3.7 打分函数与对接流程
本研究使用 DOCK3.7.2,其打分函数基于以下物理项的加和:
- • :AMBER van der Waals 势能,通过预计算能量格点(energy grid)快速评估
- • :Poisson-Boltzmann 静电势(使用 QNIFFT 求解),同样以格点形式储存
- • :配体去溶剂化惩罚,基于受体遮蔽体积(occluded surface)模型
采样策略:
- • AmpC:每个分子平均采样 4054 个朝向 × 280 个构象 = 约 113 万个构象
- • D4 受体:每个分子平均采样 3300 个朝向 × 479 个构象 = 约 158 万个构象
- • 每个构象进行刚体单纯形(simplex)能量最小化
- • 平均处理速度:1 秒/分子
计算规模:
- • D4 筛选:采样约 70 万亿个受体-配体复合物构象
- • 总计算耗时:43,563 核心小时 ≈ 1.2 个日历天(1500 个 CPU 核心并行)
2.4 蛋白受体结构准备
靶标 | PDB 结构 | 共结晶配体 | 特殊处理 |
|---|---|---|---|
AmpC β-内酰胺酶 | 1L2S | 26 μM 噻吩羧酸酯 | 增强5个关键残基部分电荷幅度(不改变净电荷);向结合位点延伸低介电区域 |
D4 多巴胺受体 | 5WIU | nemonapride | 同上低介电区域延伸;增加去溶剂化体积伪原子(radius = 0.3 Å)以改善配体电荷平衡 |
两个靶标均在正构位点(orthosteric site)计算 45 个匹配球(matching spheres),用于初始朝向采样。
2.5 聚类与多样性过滤策略
从数亿打分结果中选出测试候选物需要多层过滤:
- 1. 相似性排除:去除与 ChEMBL 已知配体 Tanimoto 相似度(ECFP4)超过阈值的分子(AmpC:Tc > 0.45;D4:Tc ≥ 0.35)
- 2. 现货库排除:去除与 350 万现货库 Tc > 0.5 的分子(确保新颖性)
- 3. Bemis-Murcko 骨架聚类:减少骨架冗余
- 4. ECFP4 最优先聚类:AmpC 取前 100 万分子以 Tc = 0.5 聚类;D4 采用混合策略(前 200 万分子 ECFP4 聚类 + 全库骨架聚类,最终产生 42 万个混合聚类)
预聚类陷阱的重要发现:若在对接前先对库进行聚类、只对代表分子对接,则最高评分分子的打分显著下降。以 D4 为例,排名前 3000 的 47 个实验活性骨架被替换为不同的聚类代表后,排名平均下滑 112 万位,仅有 2 个原始活性骨架得以保留。这证明:对全库每个分子独立对接是发现最优配体的必要条件,无法用预聚类代替。
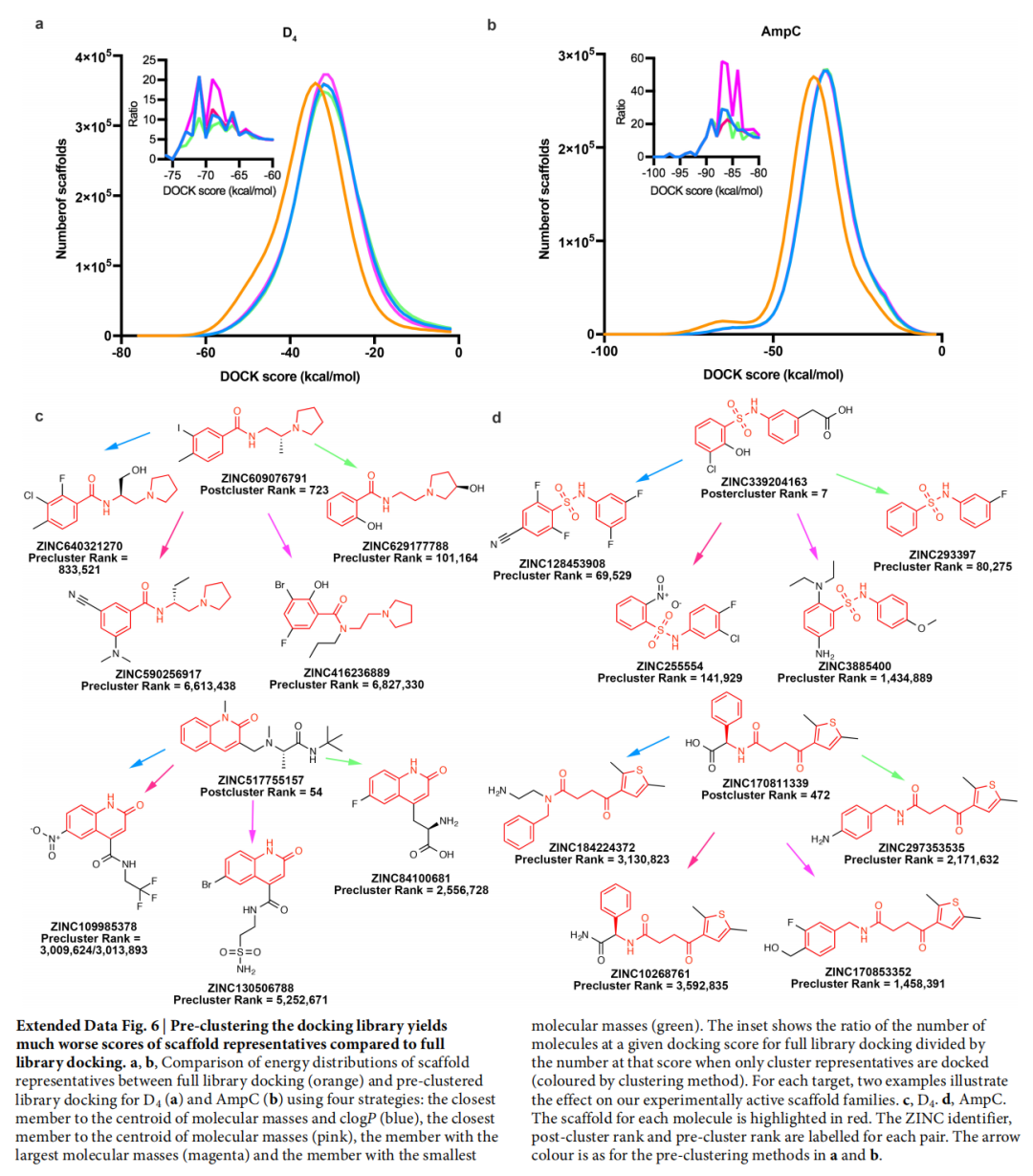
三、靶标一:AmpC β-内酰胺酶筛选
3.1 靶标背景
AmpC β-内酰胺酶(C类β-内酰胺酶)是革兰阴性菌耐药的重要机制之一,可水解大多数 β-内酰胺类抗生素。其活性位点含有催化性丝氨酸(Ser64)和"氧负离子穴"(oxyanion hole,由 Ala318-Gly317-Thr319 主链 NH 构成)。已报道的非共价 AmpC 抑制剂活性较弱(通常 Ki > 10 μM),开发有效的可逆抑制剂是长期挑战。
3.2 筛选流程与结果
一期筛选(99 百万分子 → 44 个合成测试):
- • 对接后取前 100 万分子,骨架聚类后人工挑选 51 个代表性骨架
- • 44/51(86%)成功合成
- • 5/44(11%)可测量抑制 AmpC,Ki 范围:1.3–400 μM
- • 所有 5 个命中物均为选择性竞争性抑制剂,不聚集,不抑制胰蛋白酶、糜蛋白酶和苹果酸脱氢酶
关键命中物 ZINC339204163(Ki = 1.25 μM):
这是一个含苯酚负离子(phenolate)药效团的分子。Phenolate 作为带负电荷的氧与 AmpC 氧负离子穴(通常接纳带负电荷的过渡态类似物)结合,形成三个氢键。这一作用模式在 β-内酰胺酶抑制剂中罕见;在其他酰胺酶和蛋白酶抑制剂中也鲜有记录。
3.3 类似物优化
从库内选取与 5 个初始命中物 ECFP4 Tc ≥ 0.5 或共享核心子结构的 90 个类似物,进行实验验证:
- • 超过 50% 的类似物有活性,各系列亲和力提升 3–29 倍
- • ZINC549719643(Ki = 77 nM):phenolate 系列最优类似物,是迄今已知活性最强的非共价 AmpC 抑制剂之一,比之前报道的最佳非共价抑制剂强逾 20 倍
3.4 X 射线晶体结构验证
四个新配体与 AmpC 的共晶结构被解析至高分辨率:
化合物 | PDB ID | 分辨率 | RMSD(docking vs crystal) | 备注 |
|---|---|---|---|---|
ZINC547933290 | 6DPZ | 1.50 Å | 1.30 Å | |
ZINC275579920 | 6DPY | 1.91 Å | 1.20 Å(核心)/ 1.98 Å(含末端环旋转) | 末端环无极性相互作用,构象差异可接受 |
ZINC339204163 | 6DPX | 1.90 Å | 0.98 Å | 最高精度 |
ZINC549719643 | 6DPT | 1.79 Å | 1.52 Å | phenolate 三个氢键完整再现 |
所有结构的初始 Fo-Fc 电子密度图在 2.5σ 时对配体轮廓清晰,可无歧义地确认配体构象和关键相互作用。这是对对接预测精度的直接实验验证。
四、靶标二:D4 多巴胺受体筛选
4.1 靶标背景
D4 多巴胺受体(D4R)属于 A 类 GPCR,主要表达于前额叶皮层,参与认知、工作记忆和奖赏回路调控,是精神分裂症、ADHD 和物质依赖的潜在治疗靶点。D4R 在序列和药理上与 D2R/D3R 高度同源,亚型选择性开发极具挑战。D4R 的选择性口袋由 F91²·⁶¹ 和 L111³·²⁸ 定义(上标为 Ballesteros-Weinstein 编号),这两个残基在 D2/D3 中为不同氨基酸,是亚型区分的结构基础。
4.2 筛选设计的双重目标
- 1. 新骨架发现:排除所有已知多巴胺能/5-HT/肾上腺素能配体类似物(ChEMBL,~28,000 个参考配体,Tc ≥ 0.35)
- 2. 命中率-打分曲线构建:从最高分到低分的 12 个评分区间内均匀抽样,共测试 549 个分子,这一策略前所未有
4.3 对接规模
- • 1.38 亿分子对接;约 70 万亿构象采样
- • 前 1000 个聚类代表人工挑选 124 个(检查相互作用质量、排除内张力),另自动抽取 444 个(覆盖全评分范围)
- • 589 个候选物中,549/589(93%)成功合成
4.4 活性筛选结果
初步筛选(10 μM,³H-N-甲基螺哌隆置换 > 50%):122/549(22%)呈阳性
剂量-响应(81 个化合物):Ki 范围 18.4 nM–8.3 μM,代表 81 种全新化学骨架,其中 30 种 Ki < 1 μM
功能学分类:
类型 | 代表化合物 | 效能 | D4 Ki | D2/D3 选择性 |
|---|---|---|---|---|
全激动剂 | ZINC621433143 | EC₅₀(cAMP) = 2.3 nM | 18 nM | >10,000 倍 |
全激动剂 | ZINC465129598 | EC₅₀ = 24 nM | 80 nM | >100 倍 |
全激动剂 | ZINC270269326 | EC₅₀ = 17 nM | ~500 nM | >10,000 倍 |
部分激动剂 | ZINC464771011 | EC₅₀ = 10 nM | 140 nM | >10,000 倍 |
拮抗剂 | ZINC413570733 | IC₅₀ = 5.9 μM | 130 nM | — |
拮抗剂 | ZINC130532671 | IC₅₀ = 10.8 μM | 320 nM | — |
β-arrestin 偏向激动剂 | ZINC615622500 | 无 Gᵢ 活性 | 150 nM | — |
4.5 明星化合物:ZINC621433144(180 pM D4 全激动剂)
ZINC621433143 最初作为非对映体混合物测试,EC₅₀(cAMP) = 2.3 nM。鉴于其对接构象中存在手性中心,研究者将4个非对映体独立合成并测试:
ZINC621433144((3R,4S) 构型):
- • cAMP 抑制(Gᵢ通路):EC₅₀ = 0.18 nM(180 pM)
- • Ki(D4) = 4.30 nM;Ki(D2) > 10,000 nM;Ki(D3) > 10,000 nM → 亚型选择性 > 2500 倍
- • BRET 功能偏向:Gᵢ/β-arrestin 偏向因子 = 17(与参考激动剂奎吡罗相比)
- • 是迄今报道的活性和选择性最高的 D4 受体选择性全激动剂之一
立体化学与信号偏向的关系(同一系列4个非对映体):
化合物 | 构型 | Gᵢ EC₅₀ | 偏向因子 | 方向 |
|---|---|---|---|---|
ZINC621433144 | (3R,4S) | 0.18 nM | 17 | → G蛋白偏向 |
ZINC361131264 | 另一构型 | — | 26 | → G蛋白偏向 |
ZINC361131265 | 另一构型 | — | 11 | → G蛋白偏向 |
ZINC621433143 | (3S,4S) | 2.3 nM | 0.14(≈7 倍反向) | → β-arrestin 偏向 |
单个手性中心的翻转直接反转了信号偏向方向,这是功能选择性(functional selectivity / biased agonism)领域的精彩例证,对GPCR药物设计具有重要启发。
4.6 结合模式分析
高排名分子普遍呈现以下结合特征:
- • 保守相互作用:与 D115³·³²(保守天冬氨酸,阳离子-π/盐桥)和 S196⁵·⁴²形成相互作用
- • TM5/TM6 填充:占据由 F410⁶·⁵¹ 和 F411⁶·⁵²定义的疏水区
- • D4选择性口袋利用:与 F91²·⁶¹ 和 L111³·²⁸形成相互作用(这两个残基在 D2/D3 中不同),解释了 30–500 倍的亚型选择性
- • 胞外环2氢键:部分分子与 ECL2 主链原子形成氢键,文献报道与信号偏向相关
五、命中率-打分曲线:虚拟筛选预测能力的量化验证
5.1 实验设计
将 549 个测试分子分布于 12 个打分区间(−75 到 −35 kcal/mol),每区间约 35–47 个分子,在 10 μM 浓度下统一测试置换活性(同一实验人员,相同方案)。
5.2 命中率分布
观察到命中率随对接打分呈 S 型(sigmoid)单调下降:
打分区间 | 命中率 |
|---|---|
−75 ~ −65 kcal/mol(最优区) | 22–26%(平台期) |
−65 ~ −54 kcal/mol | 从 22% 线性下降至 12% |
−54 kcal/mol(dock₅₀) | ~12%(命中率中点) |
−43 kcal/mol | ~0%(达到底部平台) |
< −43 kcal/mol | 持续 0% |
5.3 贝叶斯统计模型
使用 S 型剂量-响应方程对命中率曲线进行拟合:
参数:
- • top(最大命中率)= 24%(95% CI:约 18–30%)
- • bottom(最低命中率)= 0%(固定)
- • dock₅₀(命中率中点对应的对接能)= −54 kcal/mol
- • slope₅₀(dock₅₀ 处斜率)= −1.7% per kcal/mol
先验概率设置:
- • P(top) = Beta(α=20, β=80)(期望值约 20%)
- • P(dock₅₀) = Normal(μ=−60, σ=15)
- • P(slope) = Normal(μ=−0.2, σ=0.1)
使用 Stan 进行哈密顿蒙特卡洛(HMC)采样(4 chains × 100,000 steps,adapt_delta=0.99)获取后验分布。
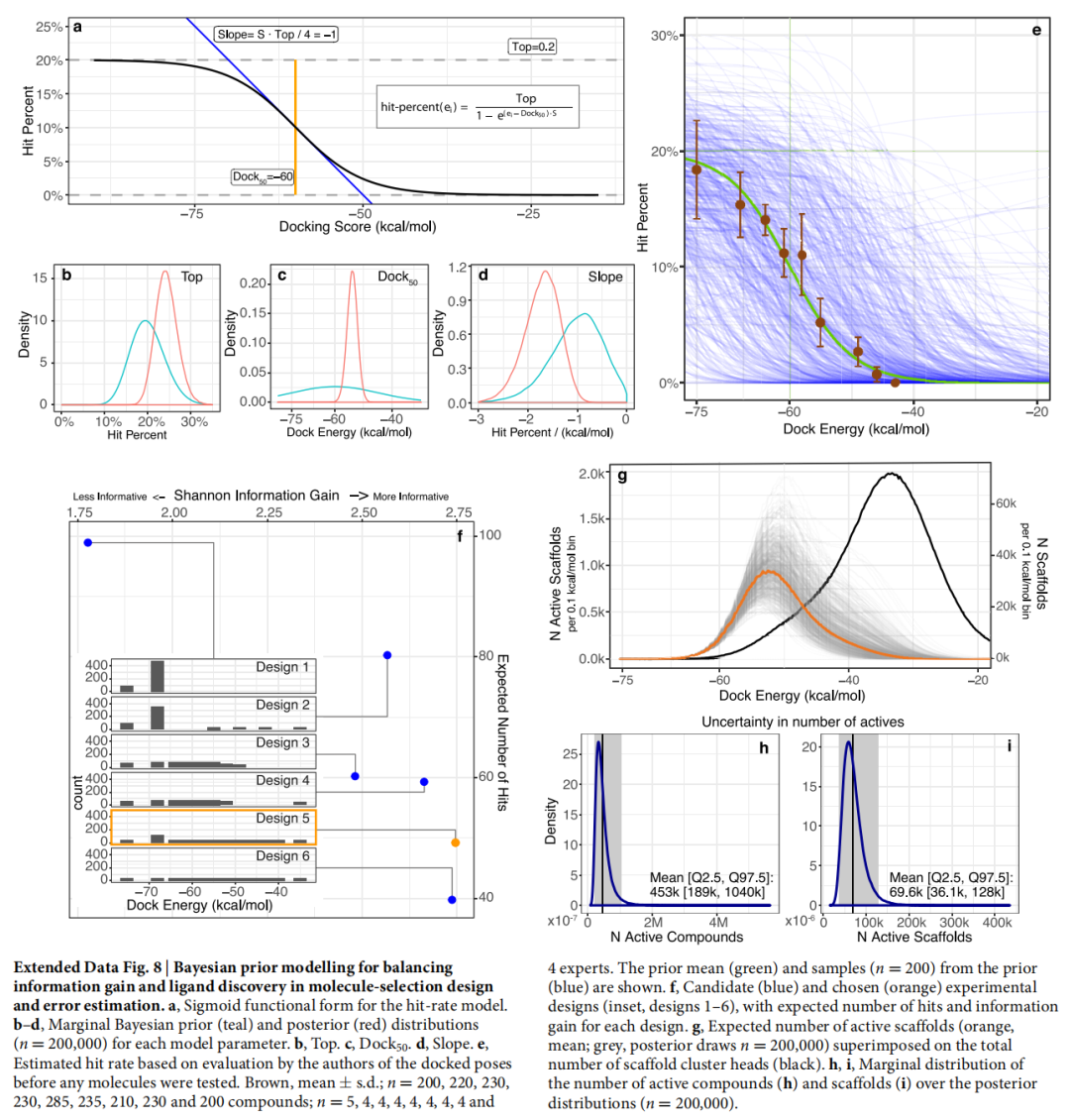
5.4 活性化合物总量估算
将命中率曲线与库内分子的打分分布积分:
估算结果:
- • Ki ≤ 10 μM 的 D4 活性化合物:453,000 个(95% CI:188,000–1,035,000),分布在 72,600 个骨架(95% CI:38,000–129,000)
- • Ki ≤ 1 μM 的 D4 活性化合物:158,000 个(95% CI:38,000–489,000)
这是首次从大规模实验数据出发,定量估算特定靶点在超大虚拟库中的活性化合物总量及骨架多样性规模。
六、人工评估 vs 自动化筛选
从前 1000 个排名聚类中,分别选取:
- • 124 个人工优选分子:对接打分 + 人工视觉评估(有利相互作用、无内张力)
- • 114 个自动选取分子:仅依据对接打分,来自相同排名区间
指标 | 人工优选 | 自动筛选 |
|---|---|---|
总体命中率(Ki < 10 μM) | ~38% | ~24% |
亚微摩尔命中率(Ki < 1 μM) | 44% | 27% |
最强激动剂 EC₅₀ | 0.18 nM(621433144) | 无 < 10 nM |
命中物的总体效能分布 | 富集高亲和力 | 均匀分布 |
结论:人机结合策略在命中率上与单纯自动化相当,但在发现高亲和力、功能活性化合物方面具有显著优势,体现了专业判断在大规模筛选中不可替代的价值。
七、关键方法论细节
7.1 虚拟库规模与对接性能的模拟研究
在前瞻性筛选之前,研究者用 DUD-E 基准集进行模拟:将已知配体与性质匹配的诱骗物(decoys)混合,模拟不同库规模下配体在前 1000 排名中的富集情况。
主要发现:
- • 对于结合位点明确的靶标(AmpC、D4、KITH 等),扩大库规模通常改善而非恶化对接性能(富集更多配体进入前 1000 排名)
- • 对于对接性能本就较差的靶标,扩大库规模可能加剧问题(噪声放大)
- • 这表明超大规模对接的前提是靶标的结合位点具有良好的三维互补性
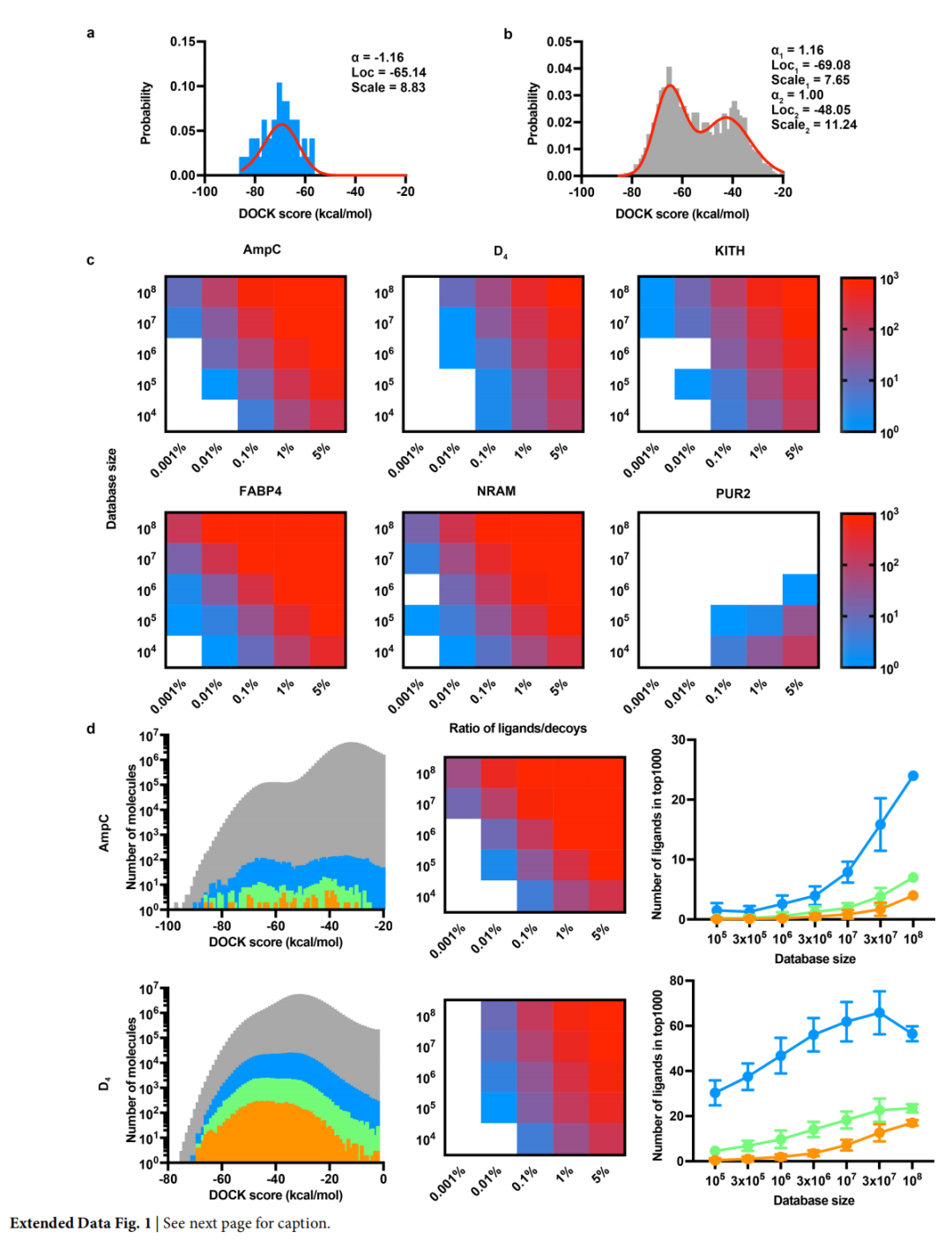
7.2 合成与质量控制
所有化合物由 Enamine 合成,采用标准双组分反应流程:
- • 交货时间:本项目中约 6 周(历史平均 3–4 周)
- • 合成成功率:本项目 93%(历史平均 85%)
- • 纯度要求:≥ 90%,大多数 ≥ 95%
- • 质量检测:LC-MS + ¹H NMR + ¹³C NMR
- • DMSO 储液:所有化合物以 30 mM 配制,测试时 DMSO 终浓度固定为 1% v/v
7.3 AmpC 酶学验证流程
- • 底物:CENTA 或头孢硝苄(nitrocephin),分光光度法检测
- • 聚集抑制剂:所有测试含 0.01% Triton X-100(防止化合物聚集假阳性)
- • 聚集验证:动态光散射(DLS)
- • 选择性验证:胰蛋白酶、糜蛋白酶、苹果酸脱氢酶三种对照酶
- • Ki 计算:Lineweaver-Burk 双倒数作图(绝大多数),少数用 Cheng-Prusoff 方程
7.4 D4 受体放射配体结合实验
- • 细胞系:HEK293T,瞬时转染 D4.4 变体(以及 D2 long、D3 用于选择性评估)
- • 放射性配体:³H-N-甲基螺哌隆(0.8–1.0 nM)
- • 初筛浓度:10 μM,孵育 2 h(室温,避光)
- • 终止方式:真空过滤(GF/A 滤膜,预浸 0.3% PEI)
- • Ki 计算:GraphPad Prism 单位点位移模型
7.5 BRET 功能偏向性检测
使用 BRET(生物发光共振能量转移)分别检测 Gᵢ 激活(Gαᵢ₁-RLuc8 / Gβγ-GFP)和 β-arrestin 招募(D4R-RLuc8 / β-arrestin-2-YFP):
- • 偏向因子计算:以奎吡罗(quinpirole)为参考激动剂,用 ΔΔlog(Emax/EC₅₀) 方法计算
- • 设备:Mithras LB940 多模微孔板读数仪
八、研究局限性
作者在讨论部分坦诚指出以下局限,值得关注:
- 1. 打分函数的系统偏差:DOCK3.7 的绝对打分值与真实结合自由能存在偏移(offset),无法可靠地对活性化合物进行精确排序,仅能提供相对优先级
- 2. 高假阳性率:即使在最优打分区间,命中率也仅约 24%——即约 76% 的高分化合物在实验中为阴性
- 3. 假阴性问题:对接必然遗漏部分真实活性化合物(构象采样不充分、打分函数偏差、氢键方向性错误等)
- 4. 命中率曲线的靶标特异性:建立的 S 型命中率曲线仅对 D4 受体经过实验验证,不能直接外推至其他靶标
- 5. 立体化学处理:许多化合物以外消旋体或非对映体混合物合成/测试,可能低估真实活性(如 621433144 在混合物中的活性被稀释)
- 6. 置信区间宽泛:估算的活性化合物总量(453,000 个)95% CI 跨越约 5 倍范围(188,000–1,035,000)
- 7. 命中率非严格单调:在最高分区间内,不同打分 bin 的命中率存在波动(22–26%),并非完全线性
九、总结评述
9.1 核心贡献
这篇论文的价值可以从以下三个层面总结:
方法论层面:在超亿级规模验证了结构对接可有效富集活性化合物,且规模的扩大不会导致信噪比的根本性退化。通过系统抽样建立的命中率-打分曲线,为虚拟筛选提供了量化的预测框架。
发现层面:在两个无关靶标上均发现了在更小库中不存在的全新化学骨架,且活性超越此前所有报道的非共价抑制剂/选择性激动剂。特别是 phenolate 系列 AmpC 抑制剂和 180 pM D4 激动剂,代表了真正意义上的"从化学空间扩展中获益"。
范式层面:通过"按需合成"模式,将可筛选的化学空间从 10⁶ 量级扩展至 10⁸–10⁹ 量级,同时保持了分子的合成可及性和质量保障。这不是对现有范式的优化,而是对药物早期发现逻辑的重构。
9.2 方法选择的深层逻辑
论文中关于"为什么不能预先聚类"的论证值得特别关注:最优配体的结构特征(决定其对接分的微观原子细节)无法在不进行实际对接的前提下预测。库内最优化合物与其聚类代表在对接打分上可能相差数十kcal/mol——这意味着对于任何高度多样化的大库,全量对接是不可绕过的技术前提。
9.3 对药物发现实践的启示
- 1. 化学空间的广度比深度更先行:初始骨架多样性直接决定了优化的天花板
- 2. 计算效能的绝对量变可引发质变:当库规模大到足以包含"理想分子"时,筛选策略的本质发生了改变
- 3. 立体化学精细控制:621433144 系列清晰展示了手性中心对效能和偏向性的精确调控,对GPCR药物设计尤其重要
- 4. 人机协同仍是最优策略:计算在广度上无可替代,而专业判断在深度和质量上仍具优势
欢迎在评论区分享你的观点~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号