BioPipelines:仅几行代码!一站式搞定蛋白/配体设计全流程

BioPipelines:仅几行代码!一站式搞定蛋白/配体设计全流程

DrugIntel
发布于 2026-03-30 15:45:21
发布于 2026-03-30 15:45:21
文献来源:Gianluca Quargnali, Pablo Rivera-Fuentes. BioPipelines: Accessible Computational Protein and Ligand Design for Chemical Biologists. bioRxiv 2026. DOI: 10.64898/2026.03.11.711024 作者单位:苏黎世大学化学系(Department of Chemistry, University of Zurich) 发表时间:2026 年 3 月 13 日(bioRxiv 预印本) 开源地址:https://github.com/locbp-uzh/biopipelines 完整文档:https://biopipelines.readthedocs.io
目录
- 1. 背景与动机:深度学习时代的工具碎片化困境
- 2. 现有框架的局限性
- 3. BioPipelines 的设计哲学与架构
- 4. 核心数据流与类型系统
- 5. 五大应用案例详解
- 6. 交互式原型与可扩展性
- 7. 与现有框架的横向对比
- 8. 批判性评估:优势、局限与展望
- 9. 总结与推荐读者群
一、背景与动机:深度学习时代的工具碎片化困境
1.1 蛋白质工程的范式转变
过去十年间,蛋白质工程经历了从"专家导向"到"民主化"的根本性转变。以 Rosetta 为代表的传统方法需要研究者深度掌握物理能量函数与复杂参数调优,门槛极高。深度学习的兴起打破了这一局面:
- • 结构预测:AlphaFold2(2021)实现了从序列到结构的近原子精度预测,此后 AlphaFold3、RoseTTAFold3(AtomWorks/RF3)、Chai-1、Boltz-2 将预测能力扩展至蛋白质-核酸-小分子多组分复合物,Boltz-2 更进一步提供结合亲和力(结合概率分类与 µM 量级回归值)预测;
- • 骨架生成:RFdiffusion、Chroma、FrameDiff 等基于扩散模型的方法使得从头设计具有条件化结构特征的蛋白质骨架成为常规操作;
- • 逆折叠:ProteinMPNN 开创了基于目标结构的快速序列设计范式,LigandMPNN 进一步将小分子结合伴侣纳入设计考量;
- • 化合物对接与分析:多种开源对接与蛋白质-配体相互作用分析工具持续涌现。
1.2 "最后一公里"问题:工具可用 ≠ 工作流可用
上述工具的突破给化学生物学带来了前所未有的机遇——从改造结合特异性、设计酶变体,到对化合物库进行靶蛋白虚拟筛选,计算设计已成为实验设计的有力前导。然而,将这些工具串联成完整设计工作流在工程层面存在严重壁垒:
挑战维度 | 具体表现 |
|---|---|
软件环境隔离 | 每个工具依赖特定版本的 Python、CUDA 及第三方库,conda 环境间几乎不可兼容 |
输入/输出格式异构 | .pdb、.cif、.sdf、SMILES、FASTA……各工具读写格式不统一,中间转换脚本易出错 |
高性能计算配置 | SLURM 作业脚本需逐工具手动编写,资源申请、依赖关系管理、失败重跑均需人工干预 |
中间文件追踪 | 多步流程产生大量中间文件,手工管理极易造成版本混乱或数据丢失 |
可复现性不足 | Shell 脚本分散、缺乏版本控制,结果难以精确复现或追溯参数 |
这些"计算后勤"问题的累积效应是:对于缺乏专职计算支持的实验型化学生物学课题组,采用计算设计的门槛往往高于科学问题本身的复杂度。
二、现有框架的局限性
在 BioPipelines 之前,学界已有若干尝试解决上述问题的框架,但均存在不同程度的局限:
ColabFold
- • 定位:通过 Google Colab 民主化蛋白质折叠工具的先驱;
- • 局限:几乎聚焦于结构预测单一环节,不支持跨工具的设计工作流编排。
Ovo
- • 定位:基于 Nextflow 工作流引擎构建的 Web 界面;
- • 局限:依赖数据库服务器与容器化基础设施,部署成本高,对实验室 IT 能力要求较高。
ProteinDJ
- • 定位:高效的多 GPU 并行蛋白质设计流水线;
- • 局限:仅提供九种预定义 pipeline 配置,不支持自定义工作流,亦不支持迭代优化。
ProtFlow
- • 定位:带集群作业管理的 Python 工具包装器;
- • 局限:
- • 缺乏面向化学生物学常见实体(蛋白质、小分子、核酸等)的类型化数据模型,模块化受限;
- • 配置代码冗长,需在整个集群运行期间维持一个持续运行的 Python 进程。
BioPipelines 明确以上述框架为竞争对手,在设计上针对性地解决了其痛点。
三、BioPipelines 的设计哲学与架构
BioPipelines 的设计围绕三个核心目标展开:抽象(Abstraction)、模块化(Modularity) 与 可测试性(Testability)。
3.1 抽象:配置与执行的两阶段分离
BioPipelines 将 pipeline 的生命周期严格划分为两个独立阶段:
阶段一:配置阶段(Python 编排)
- • 完全在 Python 中完成,利用 Python **上下文管理器(context manager)**语法定义工作流;
- • 框架在此阶段预测并生成完整的文件系统结构,自动推断每个工具的预期输入输出路径;
- • 为每个工具步骤生成自包含的 bash 脚本,负责环境激活、工具调用与输出解析;
- • 无需运行时 Python 进程:Python 脚本终止后,集群执行完全由生成的 bash 脚本驱动。
阶段二:执行阶段(bash 脚本运行)
- • 生成的 bash 脚本可独立检查、手动修改或重新提交;
- • 脚本本身既是执行产物,也是运行记录,天然具备可审计性;
- • 支持 SLURM 集群调度,资源申请(GPU 型号、时间、内存)在配置阶段一次声明。
这一设计的关键优势在于:同一套 pipeline 代码可在 Jupyter Notebook 中交互调试,无需任何修改即可提交至 SLURM 集群进行生产级运行。
3.2 模块化:标准化生物分子数据流
BioPipelines 识别出化学生物学工具操作的三种基本实体类型,将其作为工具间流通的标准数据流:
┌──────────────┐ ┌──────────────┐ ┌──────────────┐
│ Structures │ │ Sequences │ │ Compounds │
│ (.pdb/.cif/ │ │ (蛋白质/DNA/ │ │ (SMILES/CCD/ │
│ .sdf 等) │ │ RNA 序列) │ │ 等) │
└──────────────┘ └──────────────┘ └──────────────┘
↕ ↕ ↕
与各实体关联的表格数据(tabular data)每个工具以这些基本类型作为输入和输出,并附带结构化的关联数据表。例如:
- •
ProteinMPNN:Structures → Sequences - •
AlphaFold:Sequences → Structures - •
Boltz2:Sequences + Compounds → Structures(同时产出亲和力数据表)
任何新工具只需实现一个 Tool 类(包含四个方法),能够从输入流预测输出流即可被集成。框架还支持自定义数据流类型(如 Boltz2 生成的 MSA 文件可在后续运行中复用),以及基于 pandas 的标准化表格操作(过滤、排序、排名)。
3.3 可测试性:交互式执行与增量调试
- • 交互式模式:在 Jupyter/Google Colab 中运行时,框架自动检测环境并切换至"即时执行"模式,每个工具在实例化后立即运行,控制台输出实时流式显示;
- • 默认可视化:产生结构的工具(AlphaFold、Boltz2、RFdiffusion 等)自动渲染 py3Dmol 交互式 3D 查看器;图表工具内嵌显示;
- • Load 工具:可将已完成 pipeline 的任意步骤输出加载回 Jupyter,加载结果与原始输出行为一致,可直接作为后续工具的输入,无需重新运行。
四、核心数据流与类型系统
4.1 三种基本实体及其典型表示
实体类型 | 典型格式 | 示例工具 |
|---|---|---|
Structures(结构) | .pdb、.cif、.sdf | PDB 加载、AlphaFold 输出、Boltz2 输出 |
Sequences(序列) | 蛋白质氨基酸序列、DNA/RNA 序列 | ProteinMPNN 输出、DNAEncoder 输出 |
Compounds(化合物) | SMILES、CCD 代码 | CompoundLibrary 定义、Ligand 声明 |
4.2 组合控制符
BioPipelines 引入了两个组合辅助类(combinatorial assistants),用于精确控制多实体预测中的分组逻辑:
- •
Bundle:将多个实体捆绑进同一次预测(用于构建同二聚体、多链复合物等); - •
Each:对集合中的每个实体分别进行独立预测(用于库筛选、逐一评估等)。
两者可嵌套,支持复杂的组合场景(如"对每种配体,均在同一同二聚体+另一分子共同存在时计算亲和力")。
4.3 Panda 工具与表格操作
框架内置 Panda 工具(封装 pandas),提供标准化的表格操作接口:
- • 基于任意指标(pLDDT、RMSD、结合亲和力等)的过滤、排序与取前 N 名;
- •
concat(合并多步结果)、add_source(追踪数据来源)等操作; - • 与结构/序列流的无缝联动:筛选后的表格可直接驱动后续工具的输入选择。
五、五大应用案例详解
案例一:蛋白质序列重设计与基因合成(Inverse Folding + Gene Synthesis)
科学目标:在保持折叠拓扑不变的前提下,为目标蛋白生成具有改善理化性质(稳定性、溶解度)的替代序列,并输出可直接用于基因合成的密码子优化 DNA。
Pipeline 流程:
PDB("4LCD", chain="E") # 加载泛素晶体结构(chain E)
↓
ProteinMPNN(soluble_model=True, # 使用可溶蛋白训练权重重设计序列
num_sequences=10) # 生成 10 条候选序列
↓
AlphaFold(proteins=sequences) # 折叠验证,计算结构置信度
↓
DNAEncoder(organism="EC") # 输出大肠杆菌密码子优化 DNA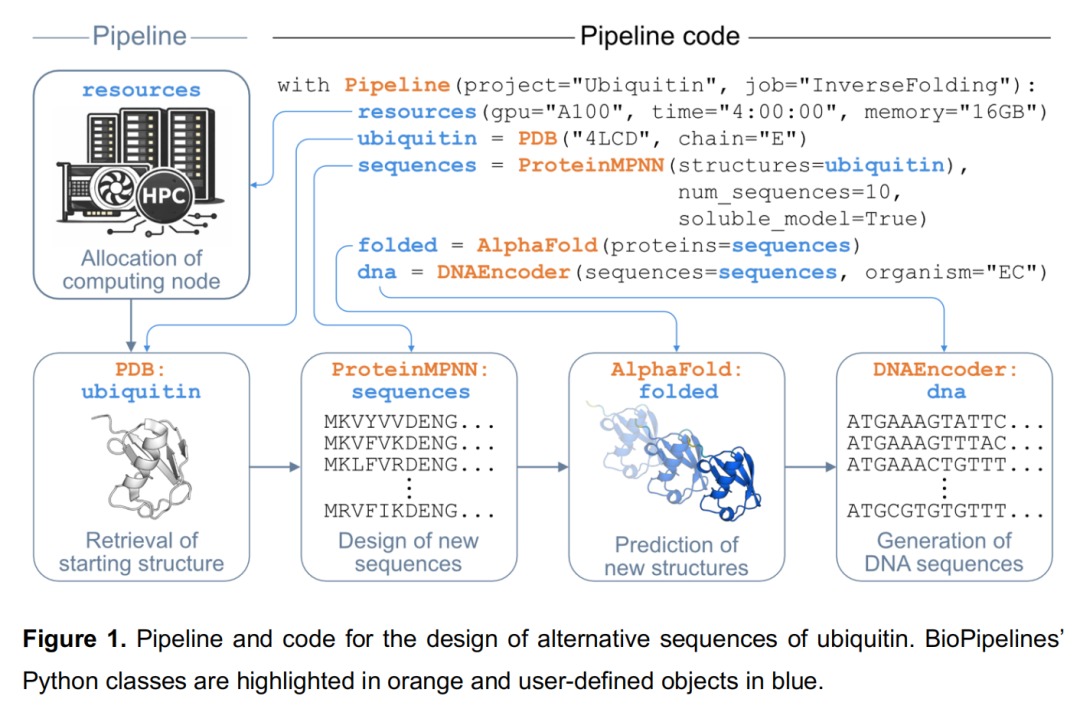
技术亮点:
- •
ProteinMPNN调用专门针对可溶蛋白训练的模型权重(soluble_model=True),相比通用模型对溶解度预测更可靠; - •
DNAEncoder调用 CoCoPUTs 数据库提供的物种特异性密码子使用频率表,采用"阈值加权采样"策略:优先使用高频密码子以避免稀有密码子,同时引入随机性以降低基因重复序列(有利于基因合成); - • 可追加
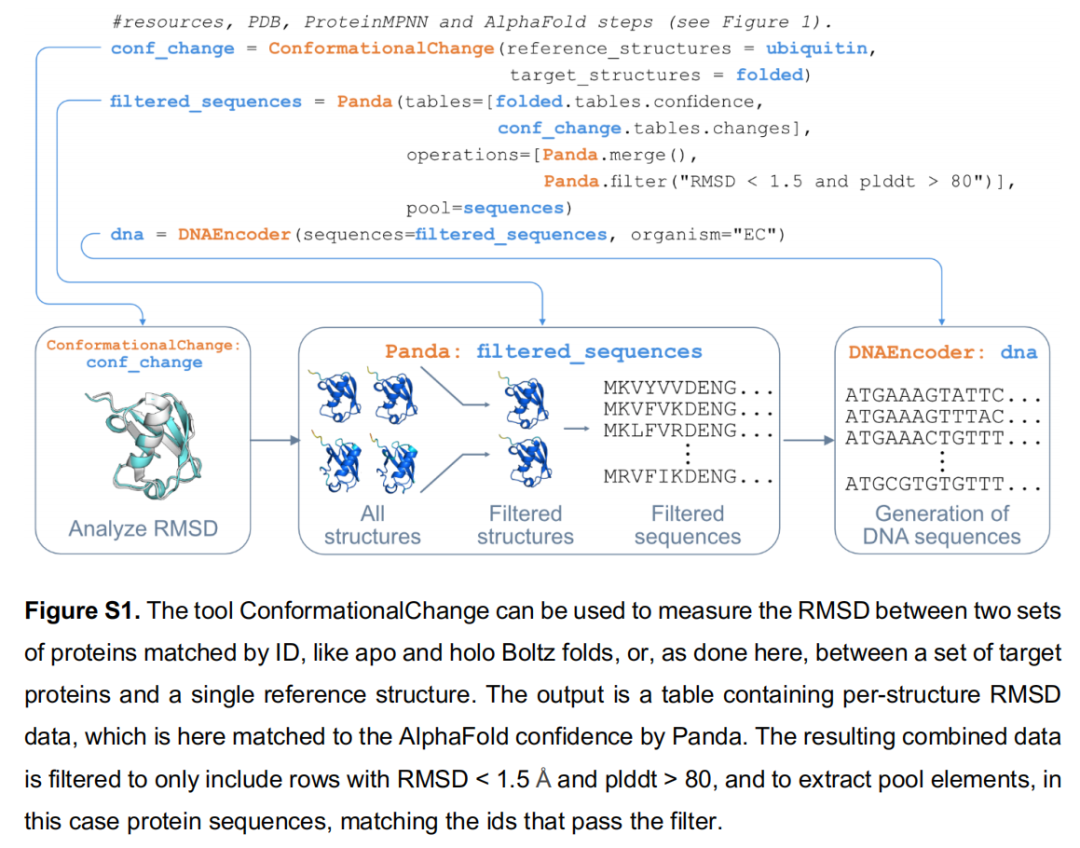
RMSD 计算 + pLDDT 阈值过滤(via Panda),对设计结果进行自动质控,仅保留与亲本结构高度相符且预测置信度高的变体(见 Figure S1)。
案例二:蛋白质结构域从头设计(De Novo Domain Design)
科学目标:替换腺苷酸激酶(adenylate kinase, PDB: 4AKE)的非必需 LID 结构域(残基 A118-160),用长度可变的全新骨架取代,同时保持其余部分结构完整。
Pipeline 流程(经典 RFdiffusion → ProteinMPNN → AlphaFold2 三步范式):
PDB("4AKE")
↓
RFdiffusion(contigs='A1-117/50-70/A161-214', # 固定非LID区域,扩散生成50-70aa新骨架
num_designs=10) # 生成10个骨架
↓
ProteinMPNN(num_sequences=2, # 每个骨架设计2条序列
redesigned=backbones.tables. # 关键:自动传递各骨架的"设计区域"信息
structures.designed)
↓
AlphaFold(proteins=sequences) # 折叠验证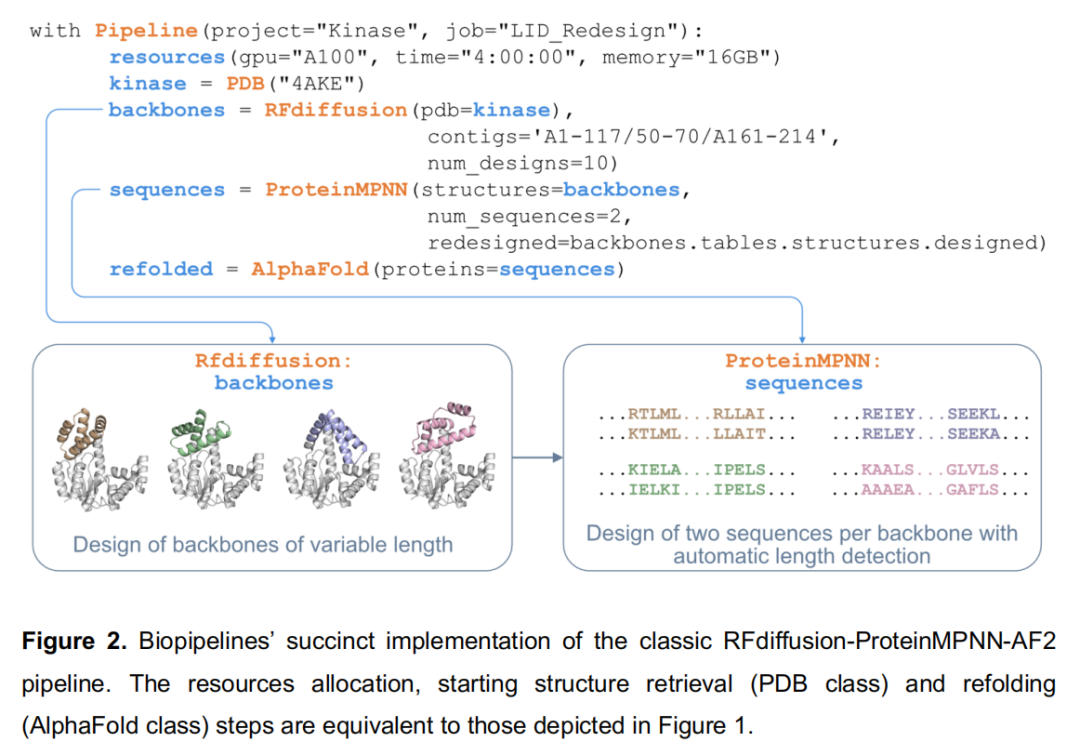
技术亮点:
- •
contigs参数中A1-117/50-70/A161-214是 RFdiffusion 的标准 contig 语法,表示固定 A1-117 和 A161-214 区域,在其间扩散生成长度为 50-70 aa 的新片段; - • 信息流自动传递:
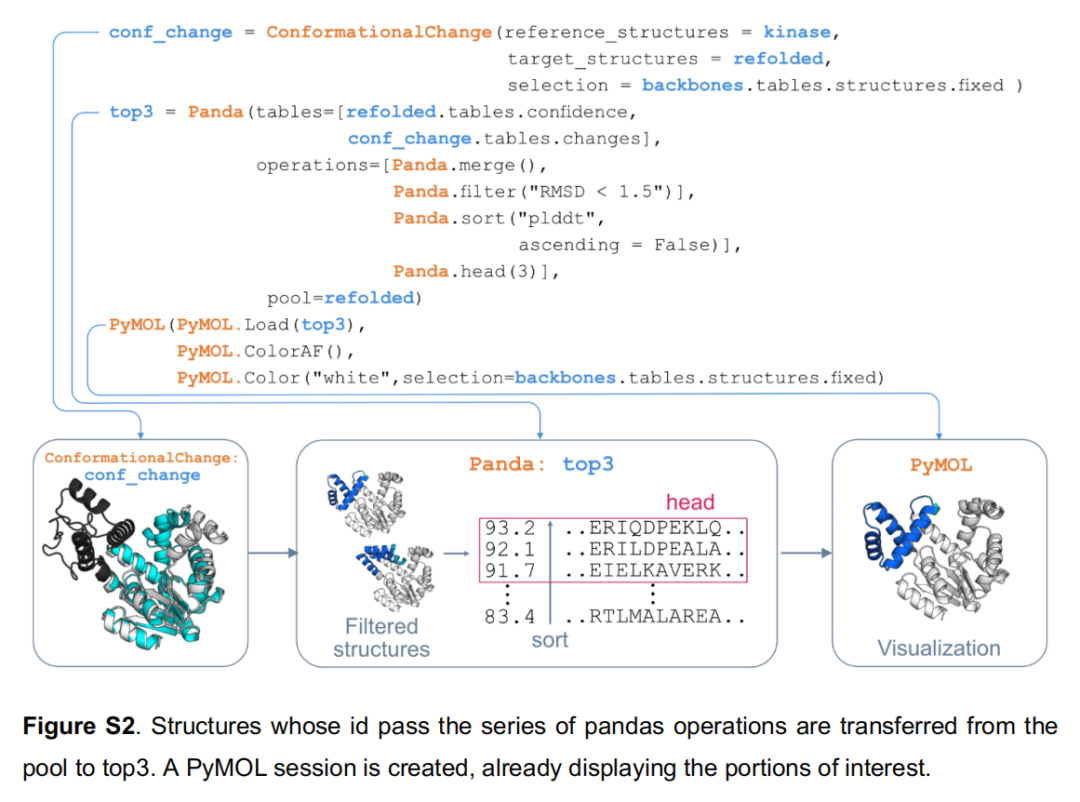
redesigned=backbones.tables.structures.designed这一参数调用是框架模块化的典型体现——RFdiffusion 在运行时通过 contig 采样确定了每个骨架中被设计的具体位置(长度可变),这一信息以结构化表格形式传递给 ProteinMPNN,后者自动仅对新生成的骨架片段设计序列,而不对固定区域进行重设计,无需任何手工解析; - • 后处理支持:可追加基于构象变化过滤(非设计区域 RMSD)→ pLDDT 排序 → 输出包含置信度着色的 PyMOL 会话文件(.pse)(见 Figure S2)。
说明:RFdiffusion 类的 pipeline 通常需要数百至数千个设计才能产出高质量命中,实际应用中
num_designs应远大于示例值。
案例三:化合物库虚拟筛选(Compound Library Screening)
科学目标:构建色氨酸衍生物小分子库,通过 Boltz2 对色氨酸阻遏蛋白(TrpR)同二聚体在其 DNA 操纵子存在下的共折叠与亲和力进行预测筛选。
Pipeline 流程:
with Pipeline(project="TrpR", job="LibraryScreen"):
resources(gpu="A100", time="8:00:00", memory="32GB")
TrpR = Sequence("MAQQSPYSAAMA...VELRQWLEEVLLKSD")
DNA = Sequence("TGTACTAGTTAACTAGTAC")
library = CompoundLibrary("path/to/TrpR_library.cdxml") # ChemDraw 组合库文件
cofolded = Boltz2(proteins=Bundle(TrpR, TrpR), # 两个TrpR链捆绑→同二聚体
dsDNA=DNA,
ligands=Each(library)) # 对库中每个配体独立预测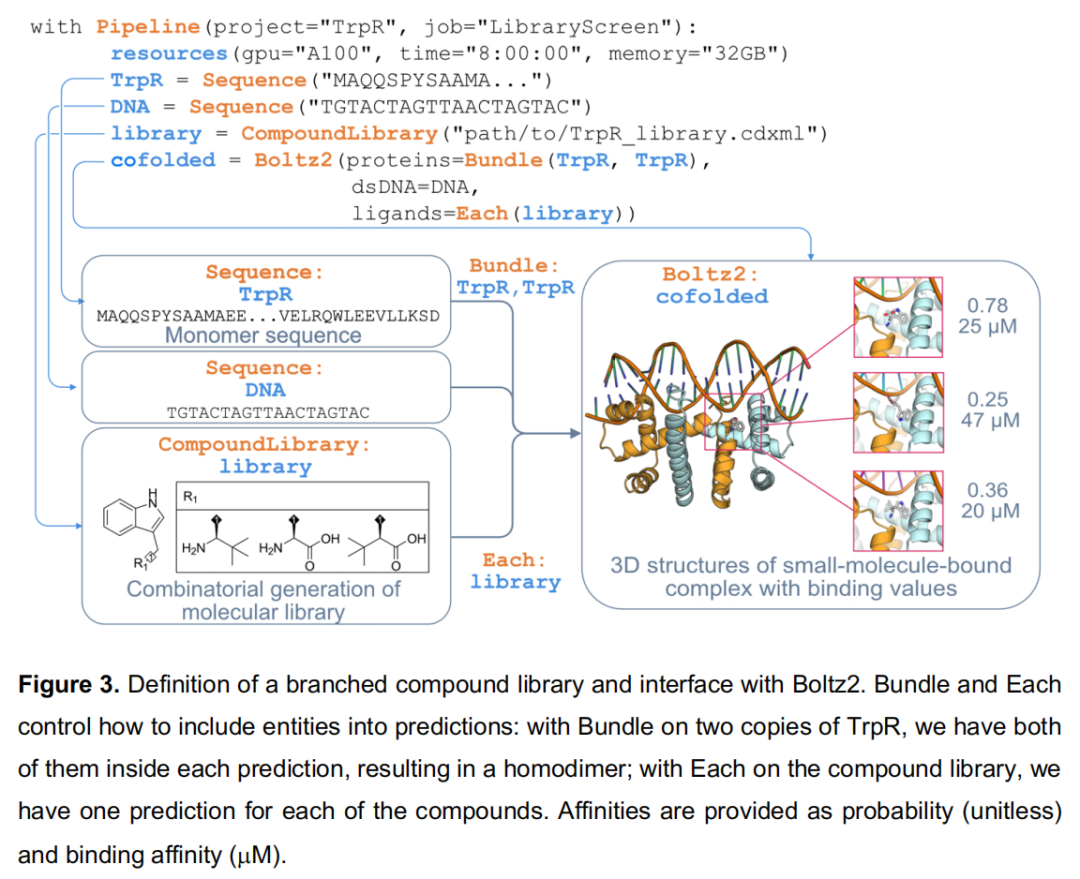
输出:
- • 每个配体的 3D 复合物结构(蛋白质-DNA-小分子三元复合体);
- • 结合概率(unitless,Boltz2 分类输出);
- • 预测结合亲和力(µM,Boltz2 回归输出)。
技术亮点:
- • 化合物库的两种生成方式:① 从带 R 基团表格的 ChemDraw (.cdxml) 文件组合生成;② 从声明了分叉点(branching points)的 SMILES 字典生成(见 Figure S3);
- •
BundlevsEach的精妙语义:Bundle(TrpR, TrpR)指示 Boltz2 将两条 TrpR 链置于同一预测中(生成同二聚体),而Each(library)指示对库中每个配体分别运行一次预测,最终得到 N 个独立的三元复合物预测; - • 结合亲和力的局限性:作者明确指出 Boltz2 的结合概率(分类)指标已有文献验证,而结合亲和力回归值"可靠性争议较大"——这一诚实的局限性声明在工具论文中较为罕见,值得注意;
- • 可追加 Plot 工具将亲和力预测值与 R 基团取代基进行关联可视化,自动导出含底层数据的 CSV 以供进一步分析(见 Figure S4)。

案例四:FRET 钙传感器融合蛋白设计(Fusion Protein / Linker Optimization)
科学目标:设计基于钙调蛋白(calmodulin)的基因编码 FRET 钙传感器,系统筛选 EBFP(供体)-钙调蛋白-EYFP(受体)三域融合蛋白的 linker 长度组合,在 apo 和 Ca²⁺ 结合态下预测结构并比较荧光基团间距。
Pipeline 流程:
with Pipeline(project="Biosensor", job="CaFRET"):
resources(gpu="A100", time="8:00:00", memory="16GB")
donor = Sequence("VSKGEELFTG...", ids="EBFP")
cam = Sequence("ADQLTEEQIA...", ids="CaM")
acceptor = Sequence("VSKGEELFTG...", ids="EYFP")
fusions = Fuse(sequences=[donor, cam, acceptor],
name="CaFRET",
linker="GSG",
linker_lengths=["0-3", "0-3"]) # 4×4=16种linker组合
apo = Boltz2(proteins=fusions) # 无Ca²⁺态
ca = Ligand("CA") # 钙离子
holo = Boltz2(proteins=fusions,
ligands=Bundle(ca, ca, ca, ca),
msas=apo) # 复用apo计算的MSA,节省服务器资源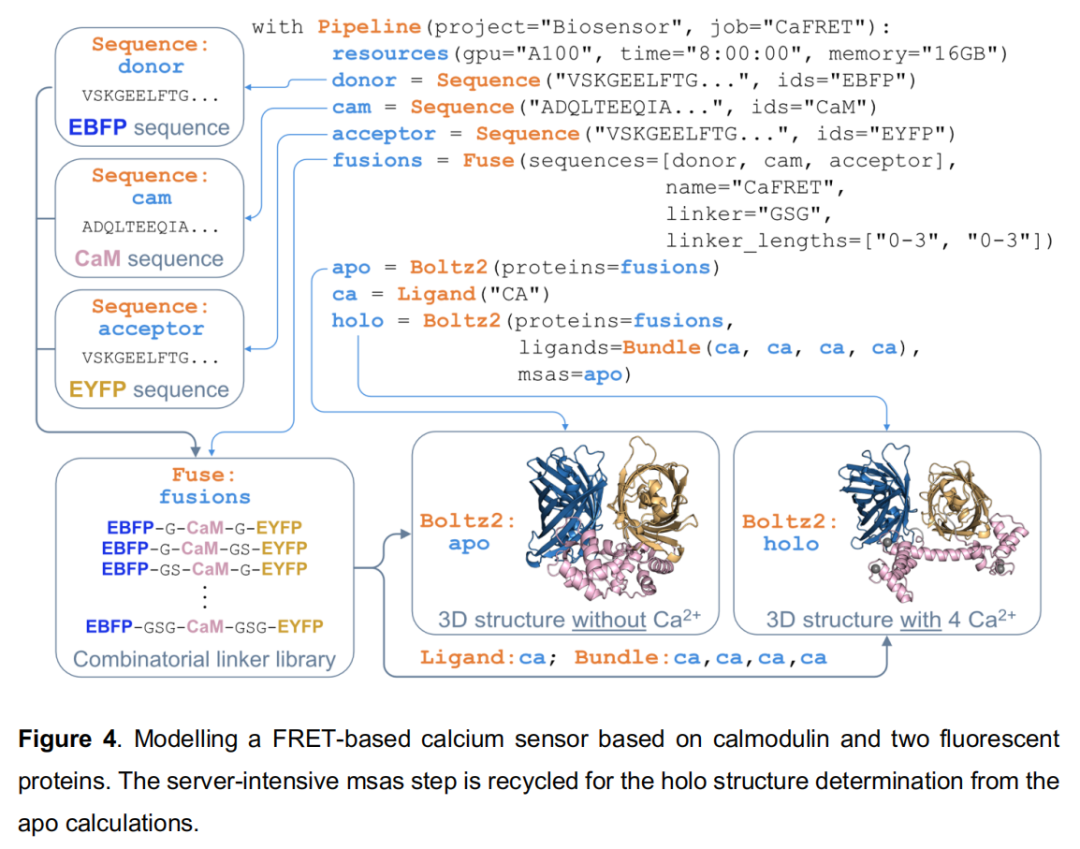
技术亮点:
- • Fuse 工具:自动生成所有 linker 长度的排列组合(4 × 4 = 16 种构型),每种构型将三个蛋白域用不同长度的 GSG linker 串联;
- • MSA 复用:
msas=apo参数将 apo 计算中已生成的多序列比对文件传递给 holo 预测,避免重复向 MSA 服务器提交计算,显著降低计算开销; - • 后续分析:Distance 工具测量供体第 66 位残基与受体 C 末端倒数第 173 位残基间的 Cα 距离(FRET 效率的核心几何参数);Angle 工具可进一步计算域间取向角;Plot 工具直接输出"哪种 linker 组合产生最大钙依赖性 FRET 几何变化"的可视化结果(见 Figures S5, S6);
- • 更复杂的 linker 探索可通过 Mutagenesis 工具结合 RFdiffusion + ProteinMPNN 实现结构化 linker 从头设计。
FRET 效率的几何假设:该示例基于荧光基团可自由旋转(取向因子 κ² = 2/3)的假设,以荧光基团间距离作为 FRET 效率的代理指标。实际传感器优化还需考虑荧光基团取向的各向异性。
案例五:迭代结合位点优化(Iterative Binding Site Optimization)
科学目标:对周质结合蛋白 NocT(PDB: 5OT9)的组氨酸(Histopine)结合口袋进行计算定向进化,多轮循环筛选亲和力更高的结合位点变体。
算法框架:
初始化:Boltz2 预测 NocT-Histopine 原始结构与亲和力
循环(5轮):
1. DistanceSelector → 识别距配体5Å以内的口袋残基
2. LigandMPNN(num_sequences=1000) → 为口袋残基设计1000条变体序列
3. MutationProfiler → 统计每个位置的突变频率(绝对频率矩阵)
4. MutationComposer(mode="weighted_random", max_mutations=3)
→ 基于频率加权采样,生成10个候选突变体(每个最多3个突变)
5. Boltz2 → 预测所有候选突变体结构与亲和力
6. Panda.sort("affinity_pred_value").head(1)
→ 选取本轮最优突变体作为下一轮模板
技术亮点:
- • 信息传递的精妙之处:
current_best = Panda(tables=[current_best.tables.result, predicted.tables.affinity], operations=[...], pool=[current_best, predicted])展示了 Panda 工具在迭代场景下的核心能力:跨轮次合并亲和力数据,自动选取最优结构作为下一轮起点,所有文件管理由框架自动处理; - •
suffix(f"Cycle{cycle+1}"):为每轮生成的文件添加循环编号后缀,保证多轮输出不相互覆盖,便于事后回溯; - • 可扩展的优化策略:文章明确指出,当前实现的是最简单的贪心筛选,框架可无缝替换为更先进的优化算法:
- • 机器学习辅助定向进化(MLDE)
- • 贝叶斯优化(Bayesian Optimization)
- • 主动学习(Active Learning)
- • 强化学习(Reinforcement Learning)
六、交互式原型与可扩展性
6.1 Jupyter 环境中的增量调试
BioPipelines 的交互式模式支持两种典型工作流:
- 1. 边运行边调试:直接在 Jupyter 中运行 pipeline,每步执行完毕即可检查中间结果(3D 结构、表格、图表),调整参数后重新运行下一步;
- 2. 加载历史结果:使用
Load工具从已完成的 HPC 运行中加载任意步骤的输出,在本地 Notebook 中继续分析或接入新工具步骤。
此外,框架利用 Python 的类型系统提供 IDE 原生支持:在 VS Code + Pylance 等现代 IDE 中,输入 RFdiffusion( 即可弹出所有参数名称、类型与文档说明;传入错误类型时,IDE 在运行前即会给出下划线警告。
6.2 AI 辅助工具扩展
BioPipelines 的一大亮点是对 AI 编程助手的系统性利用。作者报告,只需向 Claude Code(Anthropic,Opus 4.6 模型)提供 GitHub 仓库 URL 和一句话指令:
"Implement a BioPipelines tool for the repository:
<url>conforming to the existing tool standards."
Claude Code 将自动完成:
- 1. 读取仓库的安装说明、输入/输出格式、命令行接口文档;
- 2. 生成完整的 Tool 类,包含:环境安装脚本、参数校验、bash 脚本生成逻辑、输出解析器;
- 3. 输出可直接集成入框架的工具模块。
值得一提的是,BioPipelines 代码库本身有相当比例由 Claude Code(Opus 4.6)编写或重构(见 Acknowledgments),体现了"面向 AI 可读性"的代码组织策略——这反过来也使得框架代码更加规范、一致。
6.3 添加新工具的技术要求
若手动实现,一个新 Tool 类需实现的四个核心方法包括(见 Supporting Information 架构文档):
- 1. 从输入流预测输出文件结构;
- 2. 生成 bash 脚本(含环境激活、工具调用、输出转换);
- 3. 完成状态检测(用于集群作业的断点续跑);
- 4. 标准化输出格式化(结构、序列、化合物流 + 关联数据表)。
七、与现有框架的横向对比
特性 | BioPipelines | ProtFlow | ProteinDJ | Ovo | ColabFold |
|---|---|---|---|---|---|
自定义工作流 | ✅ | ✅ | ❌(9种固定) | ✅ | ❌ |
类型化生物分子数据流 | ✅ | ❌ | — | — | — |
小分子支持 | ✅ | 有限 | 有限 | 未知 | ❌ |
无需持续 Python 进程 | ✅ | ❌ | ✅ | — | — |
Jupyter 交互式调试 | ✅ | ❌ | ❌ | — | ✅(有限) |
SLURM 集群支持 | ✅ | ✅ | ✅ | ✅ | ❌ |
迭代优化支持 | ✅ | 有限 | ❌ | 未知 | ❌ |
AI 辅助工具扩展 | ✅(明确支持) | ❌ | ❌ | ❌ | ❌ |
部署复杂度 | 低 | 中 | 中 | 高 | 极低 |
开源协议 | MIT | — | — | — | — |
八、批判性评估:优势、局限与展望
8.1 核心优势
- 1. 语义明确的数据流类型系统:相比 ProtFlow 等框架仅传递文件路径,BioPipelines 的 Structures/Sequences/Compounds 类型系统使工具间接口真正标准化,从根本上保证了模块可替换性;
- 2. 两阶段分离的工程美学:配置生成 bash 脚本、脚本独立执行的设计,既解决了持续进程问题,又保持了人类可读的执行记录,这在科学可重现性方面具有显著意义;
- 3. 化学生物学特异性:明确支持小分子(SMILES、CCD、CompoundLibrary)、核酸、多组分复合物的工作流,而非仅聚焦蛋白质设计;
- 4. 组合辅助语法(Bundle/Each):以极简的语义解决了多链、多配体预测的组合爆炸问题,是该框架在 API 设计上颇具创意的贡献;
- 5. MSA 复用机制:在需要多次调用结构预测工具的场景(如 apo/holo 对比、迭代优化)中,MSA 复用可节省大量计算时间,体现了对实际使用成本的细致考量。
8.2 局限性与潜在风险
- 1. 底层模型精度依赖:框架本身是"工具的工具",最终设计质量上界取决于集成工具的精度。尤其是 Boltz2 的结合亲和力回归值,作者已明确承认其可靠性有限,在实际筛选中应以结合概率分类指标为主要参考;
- 2. HPC 基础设施依赖:SLURM 集群 + GPU 节点(示例中使用 NVIDIA A100)是生产级运行的必要条件,对缺乏 HPC 资源的课题组(如仅有本地 GPU 工作站)的支持尚不明确;
- 3. 集成工具版本漂移:计算生物学工具更新迭代快速,框架需持续维护以跟进底层工具的 API 变化。虽然 AI 辅助开发可缓解这一压力,但长期维护的可持续性仍存在不确定性;
- 4. 迭代优化算法的简化:案例五展示的是最基础的贪心单步优化,现实蛋白质工程场景(序列空间高维、适应性景观崎岖)通常需要更先进的采样策略(贝叶斯优化、主动学习等)。框架虽支持,但未提供开箱即用的实现;
- 5. 实验验证缺失:作为工具论文,所有案例均为计算展示,缺乏基于 BioPipelines 设计的实验验证结果。框架的实用价值有待下游实验工作的佐证。
8.3 与化学生物学研究实践的契合度
BioPipelines 的定位极为精准:化学生物学家既非纯实验科学家(对计算细节无感),也非计算生物学专家(无需重复造轮子),而是"有能力理解计算工具的科学逻辑,但缺乏精力处理底层工程问题"的群体。Python 上下文管理器语法与实验方案描述的高度相似性,以及内置的化合物库生成、FRET 传感器设计、基因合成输出等化学生物学特异性功能,均表明框架开发者对目标用户群体有深刻的理解。
九、总结与推荐读者群
核心贡献总结
BioPipelines 的核心贡献可概括为以下三点:
- 1. 工程层面:通过两阶段(配置/执行)分离和标准化生物分子数据流,将 30+ 个深度学习设计工具整合为一个类型安全、可组合、无需持续进程的 Python 框架,同一代码支持 Jupyter 交互调试与 SLURM 生产运行;
- 2. 科学层面:系统覆盖了化学生物学核心计算需求——逆折叠、从头设计、化合物库筛选、融合蛋白 linker 优化、迭代结合位点优化——并提供了开箱即用的案例实现;
- 3. 生态层面:通过与 AI 编程助手(Claude Code)的深度整合,显著降低了框架扩展的门槛,构建了一个具备自我生长能力的工具生态。
推荐读者群
读者类型 | 推荐理由 | 建议阅读重点 |
|---|---|---|
以实验为主的化学生物学课题组 | 核心目标用户,可大幅降低计算设计采用门槛 | 所有应用案例 + GitHub README |
计算化学/计算生物学研究者 | 了解工具整合方案,借鉴模块化架构设计 | 软件设计部分 + Supporting Information 架构文档 |
结构生物学家 | 逆折叠 + 结构预测工具链的简洁实现参考 | 案例一、二 |
药物化学家/CADD 研究者 | 化合物库筛选与 Boltz2 集成方案 | 案例三 |
合成生物学家 | 从头设计 + 基因合成一体化 pipeline | 案例一、二 + DNAEncoder 工具 |
计算工具开发者 | AI 辅助框架扩展范式,具有方法论价值 | 扩展框架部分 + Supporting Information AI 工具开发记录 |
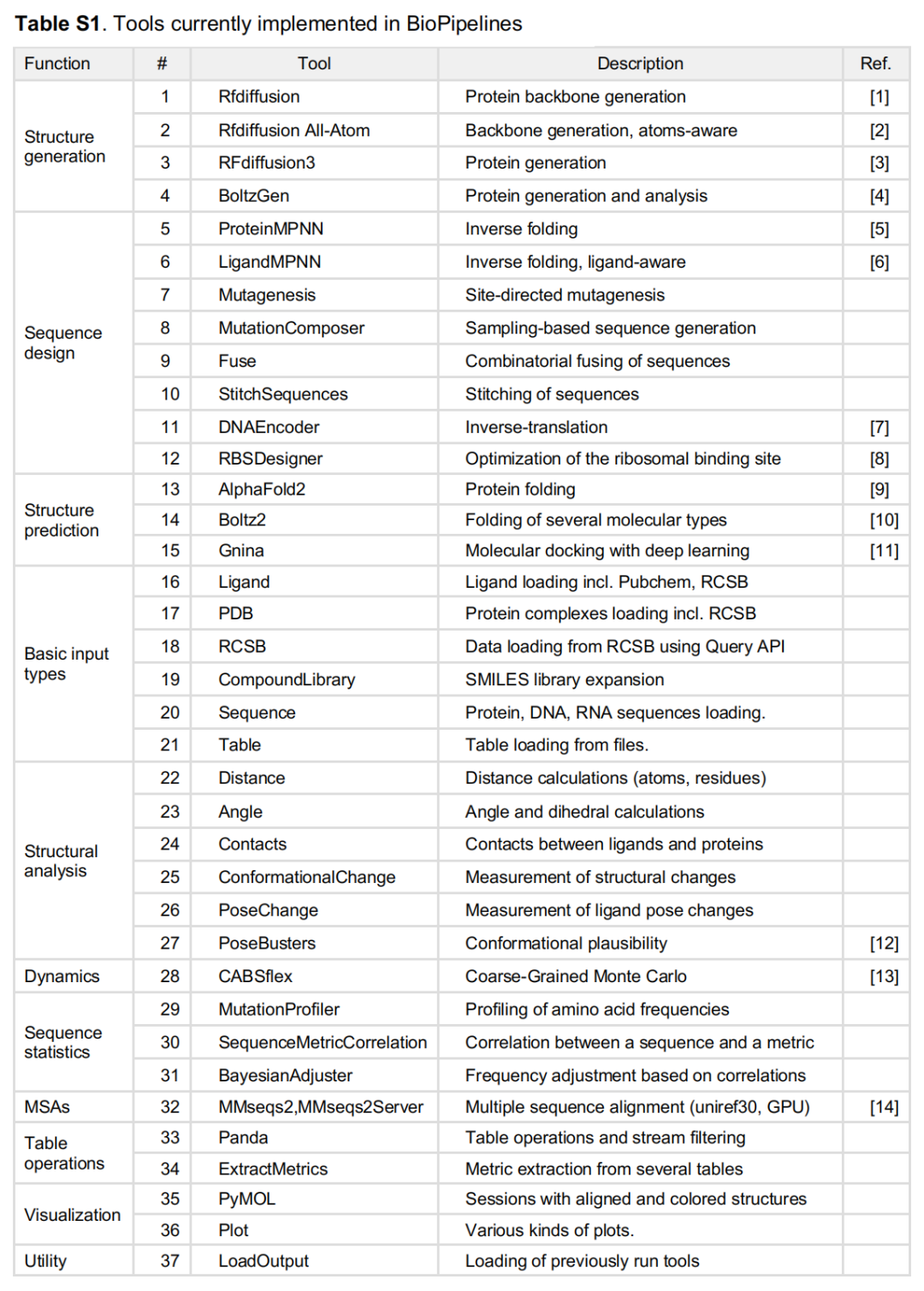
附录:框架已集成工具清单
根据论文正文及 Table S1,BioPipelines 集成的工具(>30 个)按功能分类如下:
本文为基于bioRxiv 预印本的学术解读。整理时间:2026 年 3 月
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号