Nat. Commun. | 通过深度学习偏置不均衡分布以探索化学与催化

Nat. Commun. | 通过深度学习偏置不均衡分布以探索化学与催化

DrugAI
发布于 2026-03-03 17:36:13
发布于 2026-03-03 17:36:13
DRUGONE
化学反应和催化机理的自动发现仍是计算化学中的核心难题,尤其在复杂体系中,传统方法往往难以确定有效搜索方向。研究人员提出了一种名为 Loxodynamics 的深度学习驱动反应探索方法,通过分析局部概率分布的偏斜性来自动确定最低能垒方向,并在分子动力学模拟中施加定向偏置以推动体系进入新的亚稳态。该框架的核心是 Skewencoder,一种带有偏斜损失函数的自编码器,用于从有限采样数据中提取反应坐标。通过迭代采样与搜索循环,该方法能够在有限温度条件下自适应构建自由能面,并成功应用于模型势、气相反应以及真实催化体系。该方法无需预先指定集体变量或提高温度即可加速反应探索,从而为复杂反应网络发现提供系统化工具。

从第一性原理预测化学反应路径是计算化学长期以来的重大挑战。传统流程通常依赖化学直觉猜测产物或中间体,再通过几何优化寻找最低能量结构,但这种方式高度依赖先验知识且难以处理复杂体系。另一类方法通过势能面曲率寻找反应路径,虽然在小体系中有效,但往往忽略有限温度下的构象空间。分子动力学能够考虑温度效应,但普通MD时间尺度有限,难以捕获跨越高能垒的稀有事件。为此出现了多种增强采样技术,例如升温或在集体变量方向施加偏置势,但这些方法仍需要事先设计反应坐标或已知状态。研究人员因此希望开发一种能够自动构建集体变量并同时确定最佳偏置方向的反应探索方法。
方法
研究人员提出的Loxodynamics方法首先在反应物态附近进行短时间采样,并估计局部概率分布。通过计算该分布的偏斜性,模型判断沿哪个方向移动更可能跨越最低能垒进入新状态。为将该思想推广到高维分子体系,研究人员构建了Skewencoder自编码器,将高维描述符压缩为一维潜空间集体变量,同时在损失函数中加入偏斜项,使潜空间分布最大化偏斜度。随后在该潜空间中施加半谐势壁,引导分子动力学沿预测方向推进,并在每轮迭代中更新模型。整个流程通过反复“采样—学习—施加偏置”实现自动反应探索。
结果
方法原理验证与简单势能测试
研究人员首先在一维双阱势和二维模型势上测试方法。结果显示,通过分析局部分布偏斜性,算法能够识别正确的跃迁方向,并逐步推动体系从一个势阱移动到另一个势阱。在二维势能面中,方法能够在存在不同能垒路径时自动选择最低能垒通道,并逐步扩展采样区域。这说明局部统计非对称性确实能够反映潜在反应方向,并可作为自动反应坐标构建的依据。
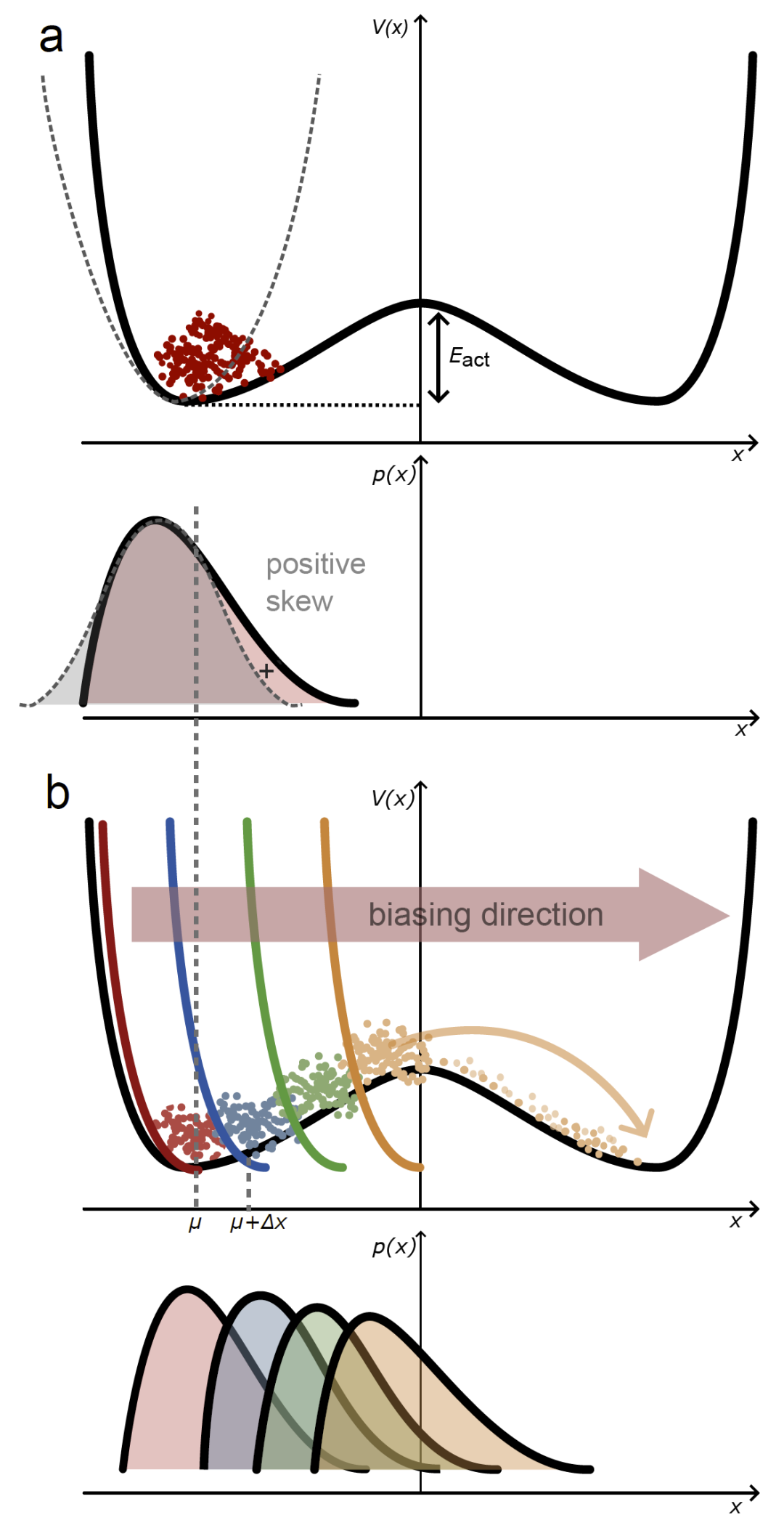
图1:Loxodynamics方法在一维情况下的示意图。
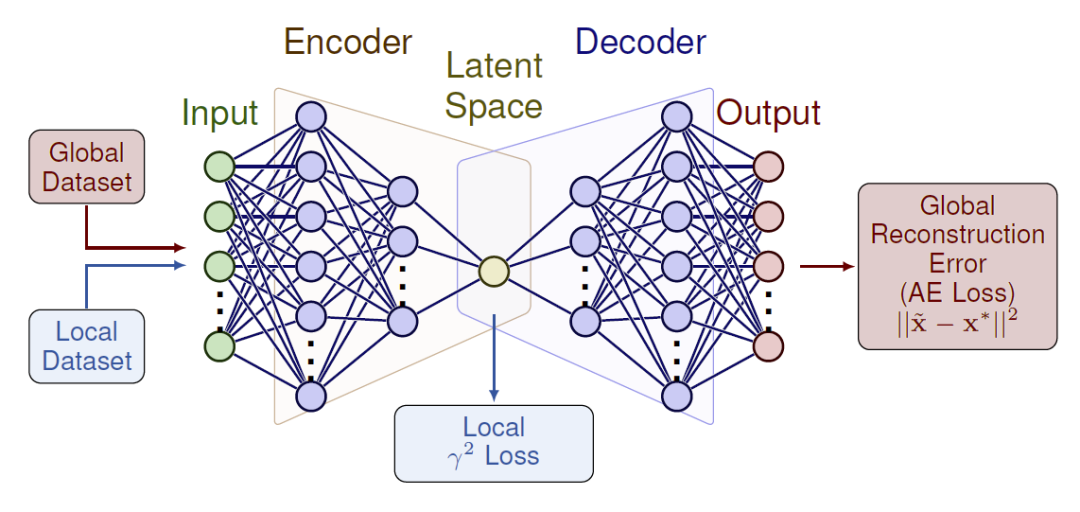
图2:Skewencoder架构示意图——一种基于单瓶颈潜空间神经元的多任务自编码器框架。
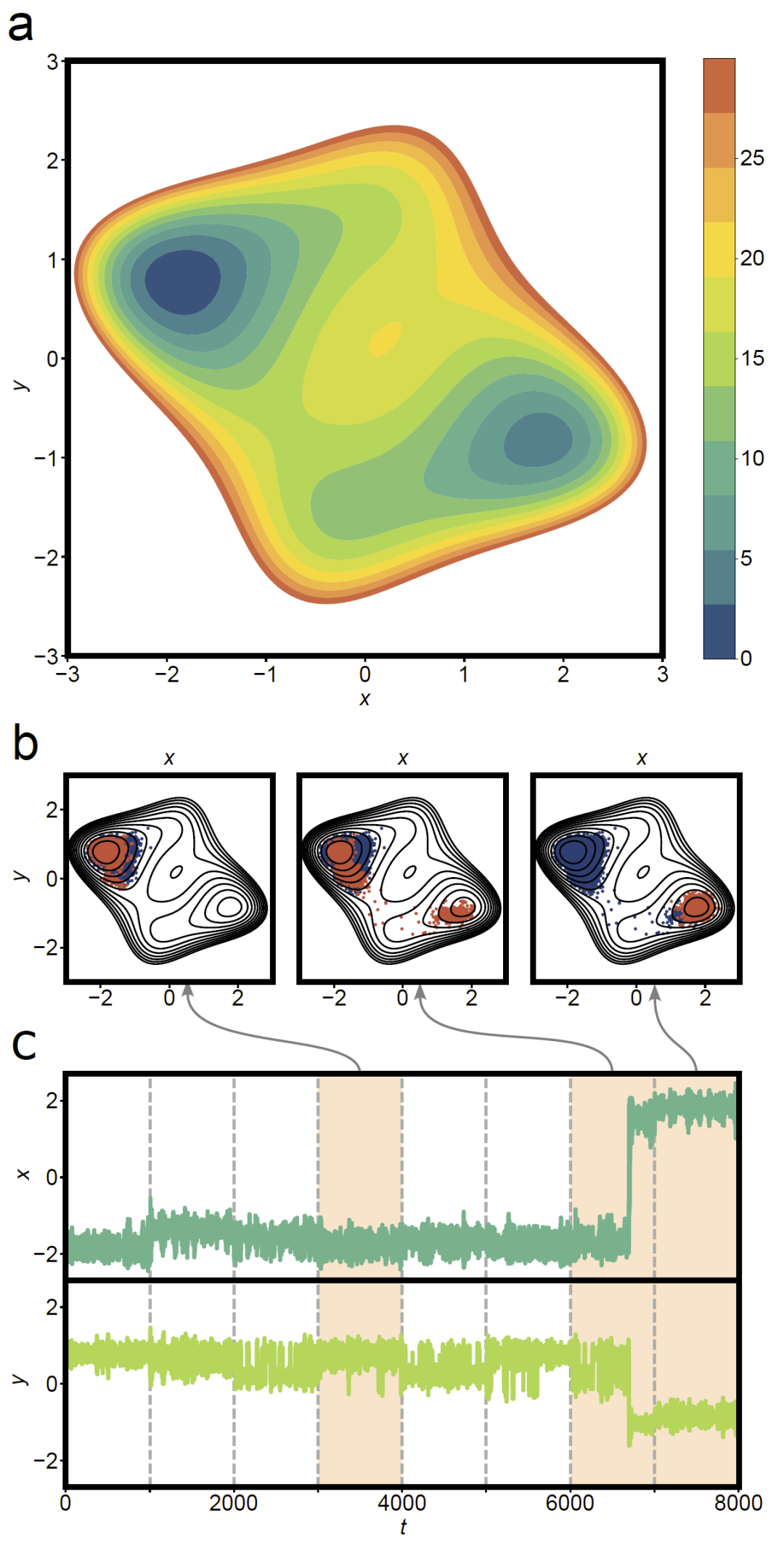
图3:二维势能面(PES)上的Loxodynamics探索过程。
气相典型反应验证
随后研究人员在两个经典气相反应上验证方法,包括SN2取代反应和Diels–Alder环加成反应。对于SN2反应,模型利用关键原子间距离作为描述符,在数轮迭代后成功识别最优方向并推动体系进入产物态。对于更复杂的Diels–Alder反应,研究人员甚至输入所有重原子间距离而未预选关键反应坐标,方法仍能自动识别反应路径并在正反方向均成功跨越能垒。这表明该方法在缺乏机制先验信息时仍具有稳健性。
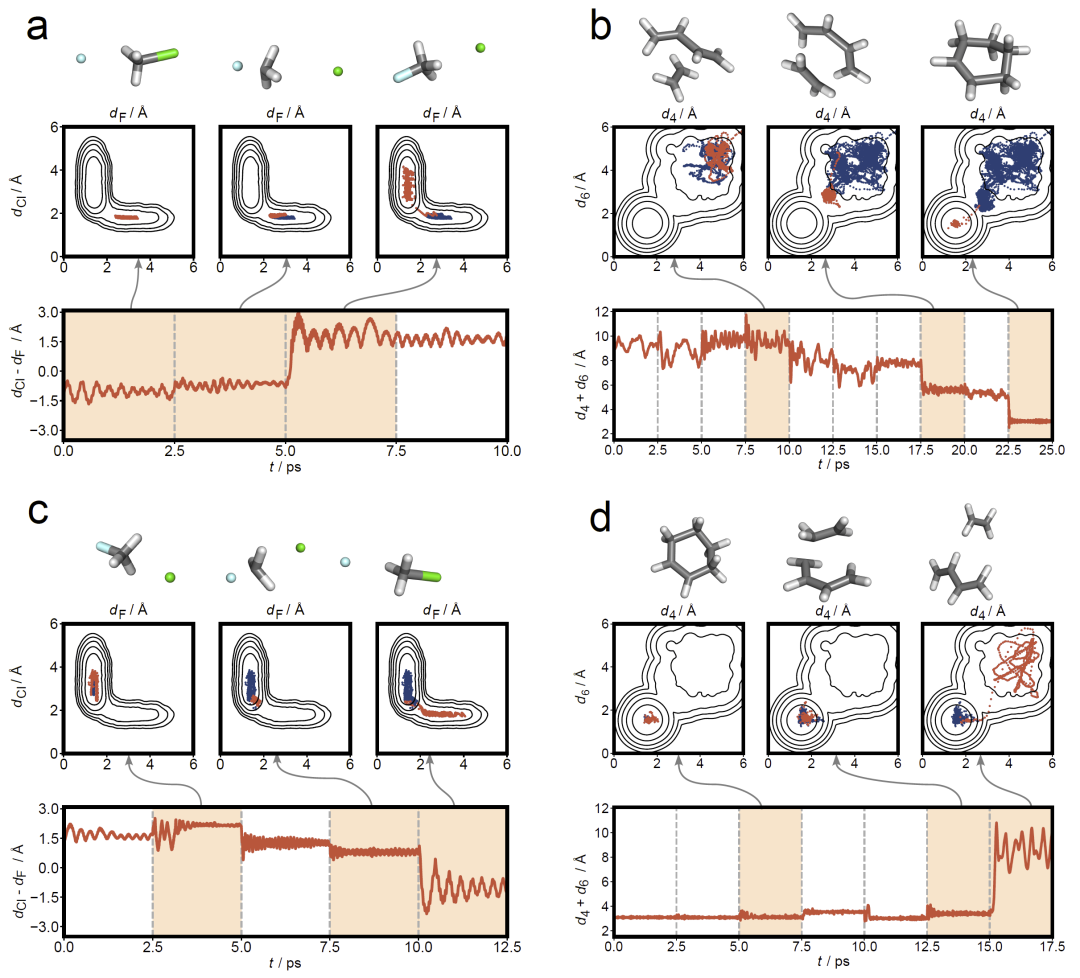
图4:两种化学反应在Loxodynamics过程中的体系演化。
真实催化体系:乙醇脱水反应
研究人员进一步将方法应用于酸性沸石中乙醇脱水生成乙烯的催化反应。该体系存在两种已知机理:协同机理和逐步机理。使用包含多种原子距离的通用描述符后,Loxodynamics不仅成功找到最终产物态,还自动发现了逐步机理中的乙氧基中间体,并进一步从该中间体继续搜索得到最终产物。结果与已有计算研究一致,证明该方法能够在复杂催化环境中同时识别产物和关键中间体。
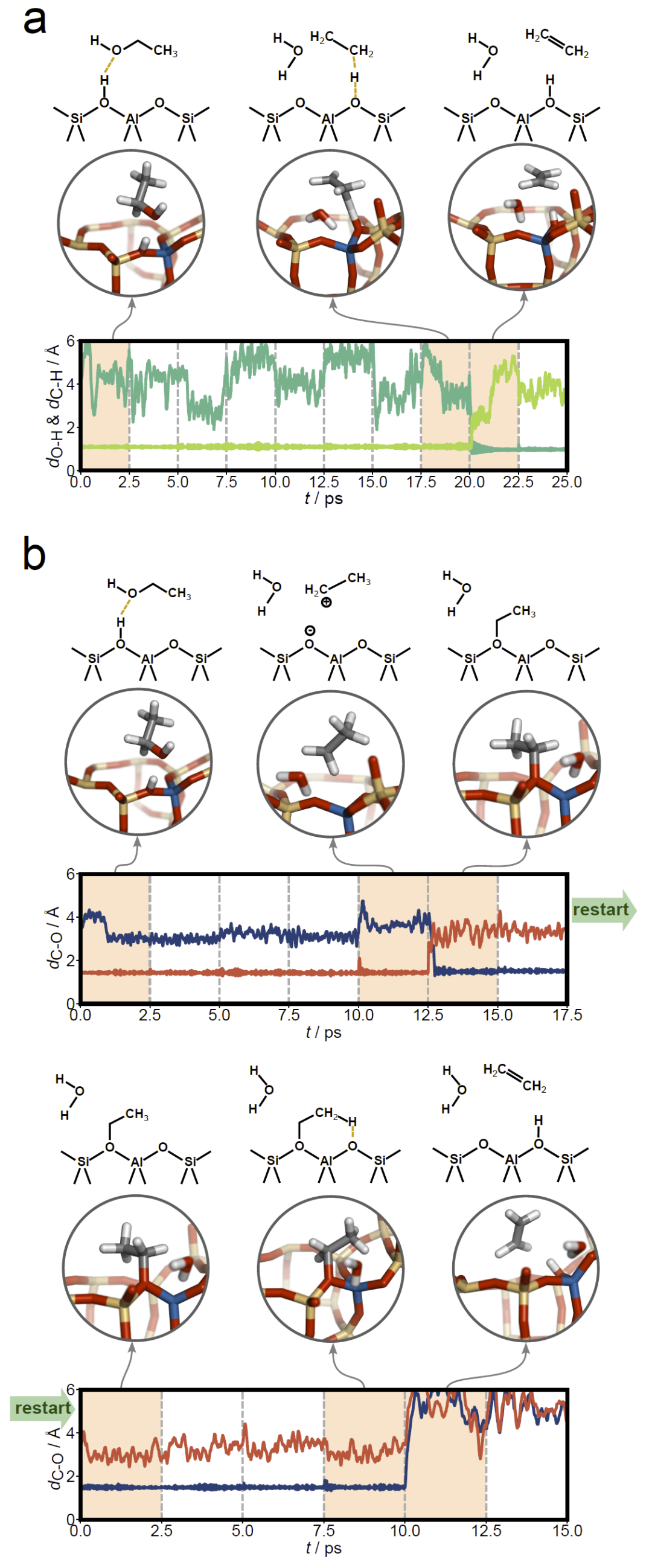
图5:酸性CHA沸石(H-CHA)中乙醇脱水反应的结构变化与原子对距离演化。
更复杂体系:丁醇脱水反应网络
在更复杂的1-丁醇脱水反应中,体系可能生成多个烯烃异构体。研究人员同样从吸附态开始探索,仅使用简单的距离描述符。结果显示模型不仅识别出1-丁烯,还找到顺式和反式2-丁烯产物,并发现丁氧基中间体。与传统需要大量人工猜测结构的DFT路径搜索相比,该方法能自动构建完整反应网络,显示出在复杂催化体系中探索多产物空间的潜力。
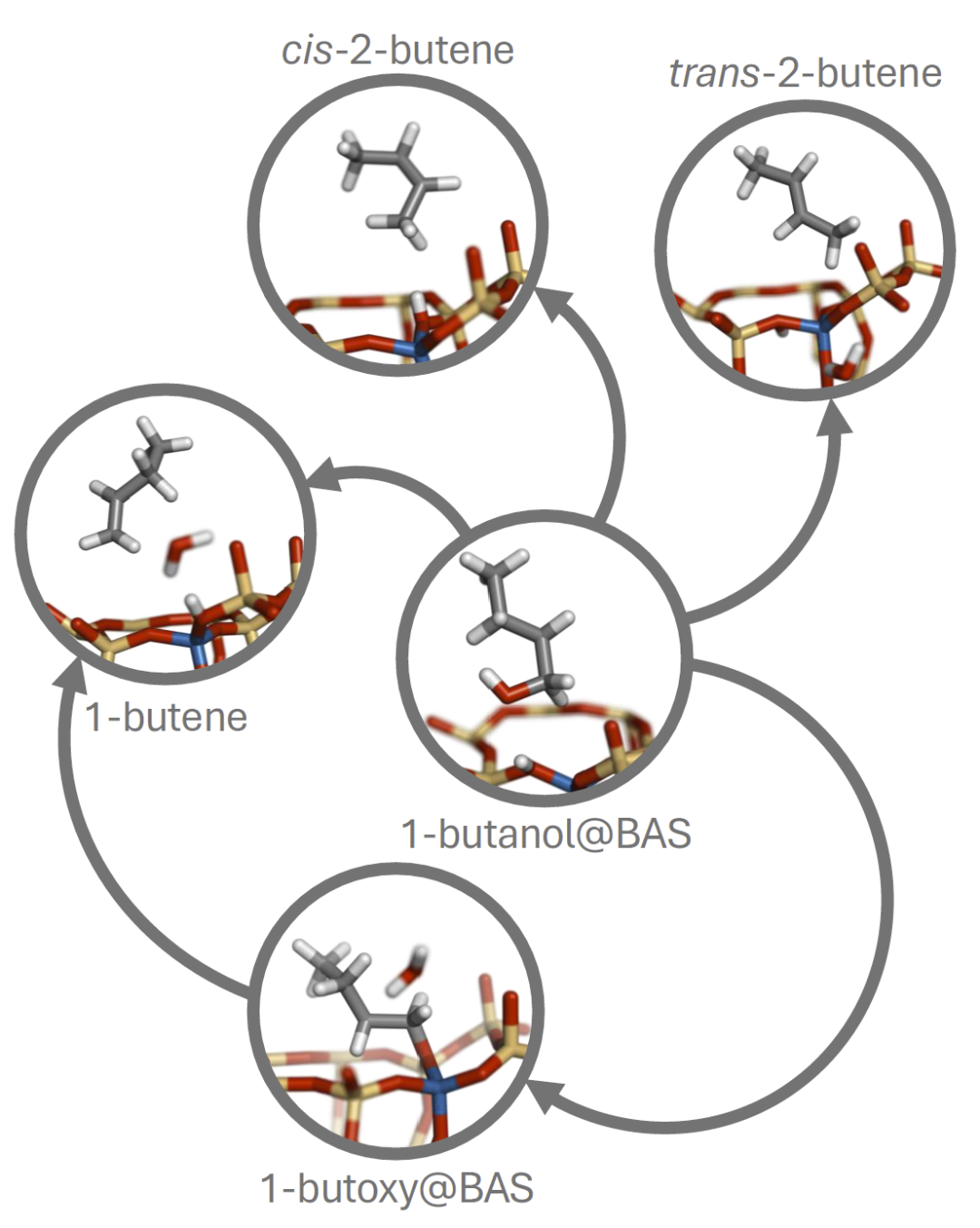
图6:Loxodynamics识别的酸性CHA沸石中1-丁醇脱水反应网络。
讨论
研究人员认为该方法的核心优势在于利用局部概率分布的不对称性来驱动反应探索,从而在有限温度下自然考虑构象波动和动力学效应。其模块化设计允许与现有分子动力学程序直接结合,并适用于多种化学与材料场景,包括表面催化、电催化、酶催化以及未知反应路径体系。该方法还可与机器学习原子势模型结合,通过主动探索转变区域来提高模型训练质量。当前实现仍需要一定程度的人工设置,例如状态识别与约束参数,但未来可通过自动代理系统进一步实现全流程自动化,从而推动复杂反应网络的自主发现。
整理 | DrugOne团队
参考资料
Zhang, Z., Piccini, G. Exploring chemistry and catalysis by biasing skewed distributions via deep learning. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-69586-8

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-02-22,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号