Cancer Cell | 癌症治疗中KRAS抑制剂的新兴格局

Cancer Cell | 癌症治疗中KRAS抑制剂的新兴格局

DrugIntel
发布于 2026-02-04 14:23:05
发布于 2026-02-04 14:23:05

作者:Riedl, Jakob M等( 原载《Cancer Cell》 2026年)
摘要
KRAS、NRAS和HRAS的变异发生在大约20%的癌症患者中,这使得RAS成为被研究最深入的致癌靶点之一。突变选择性KRAS G12C抑制剂的发现为RAS定向疗法提供了概念验证,预示着RAS驱动癌症治疗的新时代。然而,第一代KRAS G12C抑制剂的疗效因耐药性的快速出现而受到限制。具有不同作用机制和更广靶点覆盖范围的新型(K)RAS抑制剂有望克服耐药性,并将RAS靶向治疗的益处扩展到更广泛的患者群体。在本篇综述中,作者总结了KRAS G12C抑制剂跨肿瘤类型的临床证据,并描绘了关键的耐药机制。作者进一步讨论了快速发展的下一代(K)RAS抑制剂格局,特别关注其靶点选择性、作用机制、初步临床疗效,以及每类抑制剂固有的治疗机遇与挑战。
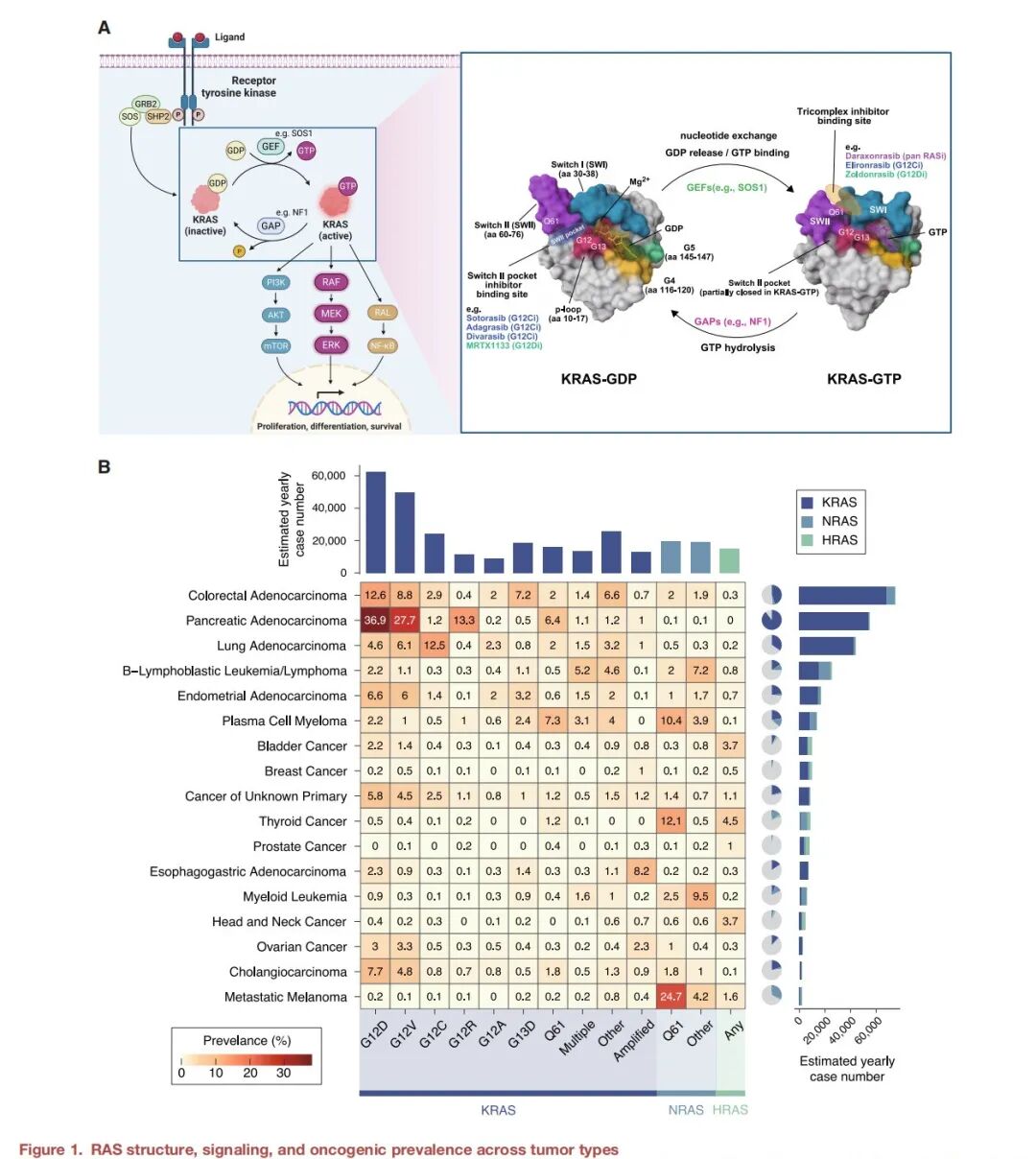
图1. RAS的结构、信号传导及其在不同肿瘤类型中的致癌突变发生率(A)RAS信号通路及与GDP/GTP结合的KRAS结构,图中标明了常见的突变位点。 图中显示了KRAS的结构基序(P环[残基10-17]、开关I区[30-38]、开关II区[60-76]、G4[116-120]、G5[145-147])以及常见的致癌突变位点(G12、G13和Q61)。KRAS在无活性的GDP结合("关闭")状态和有活性的GTP结合("开启")状态之间循环转换,该过程受鸟嘌呤核苷酸交换因子(GEFs,如SOS1)和GTP酶激活蛋白(GAPs,如NF1)调控。GTP结合的KRAS会激活下游通路,包括RAF-MEK-ERK和PI3K-AKT,从而驱动细胞增殖和存活。开关II口袋抑制剂(如sotorasib, adagrasib, divarasib, MRTX1133)靶向GDP结合的KRAS;而三元复合物抑制剂(如daraxonrasib, elironrasib, zoldonrasib)则通过结合开关I和II区之间的空隙来靶向GTP结合的KRAS。(B)不同KRAS突变和扩增在多种癌症类型中的发生率。 该热图基于AACR项目GENIE的数据(分析了108,509份患者样本),显示了KRAS、NRAS和HRAS突变在代表性癌症类型中的相对频率(%)。X轴表示RAS亚型(KRAS、NRAS、HRAS),Y轴列出了选定的癌症类型。热图上方的条形图显示了每种突变在美国的预估年度患者数,该数据结合了2025年美国癌症发病率统计和GENIE数据库得出的突变频率。右侧的堆叠条形图描述了每种癌症类型中携带KRAS、NRAS或HRAS突变的美国预估年度患者数。饼图总结了每种癌症类型内部不同RAS亚型突变所占的比例。详细分析方法见补充材料(文档S1)。
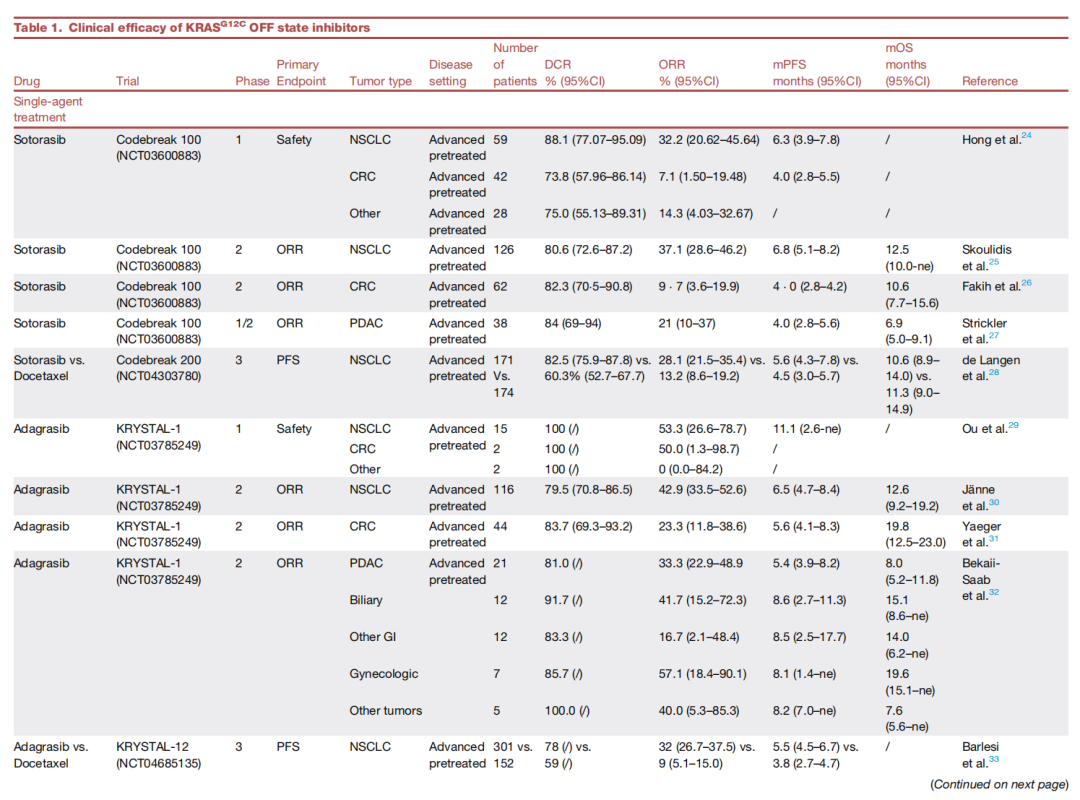
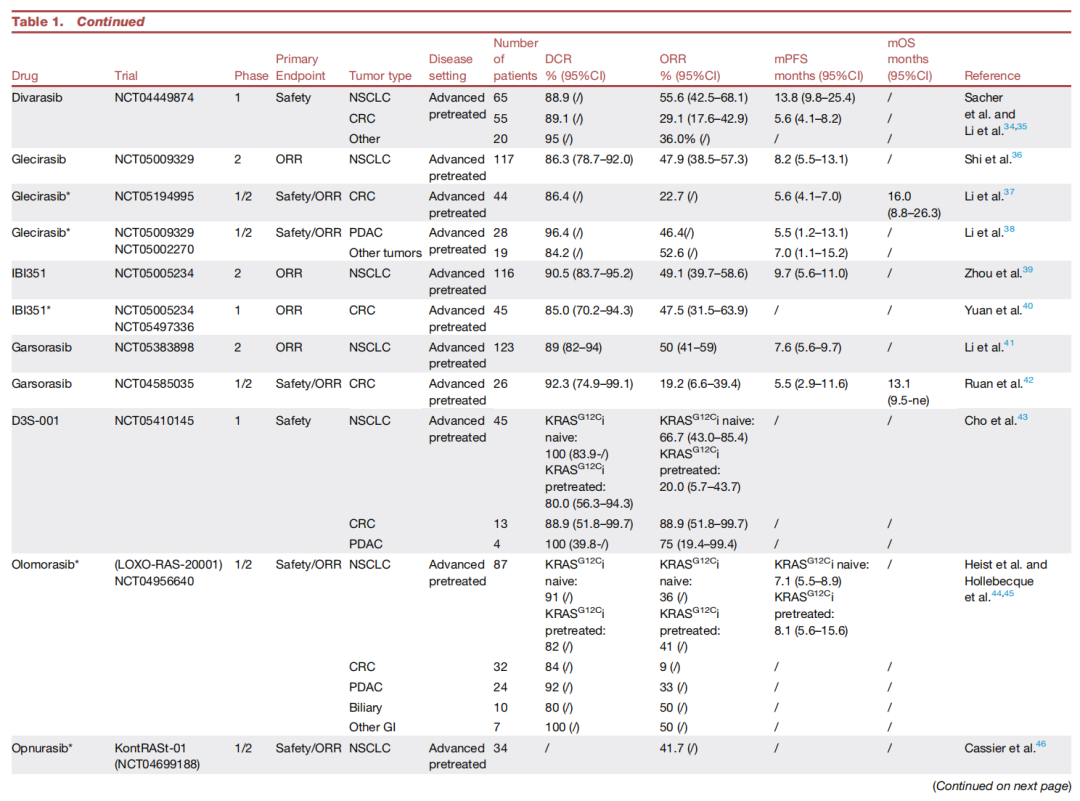
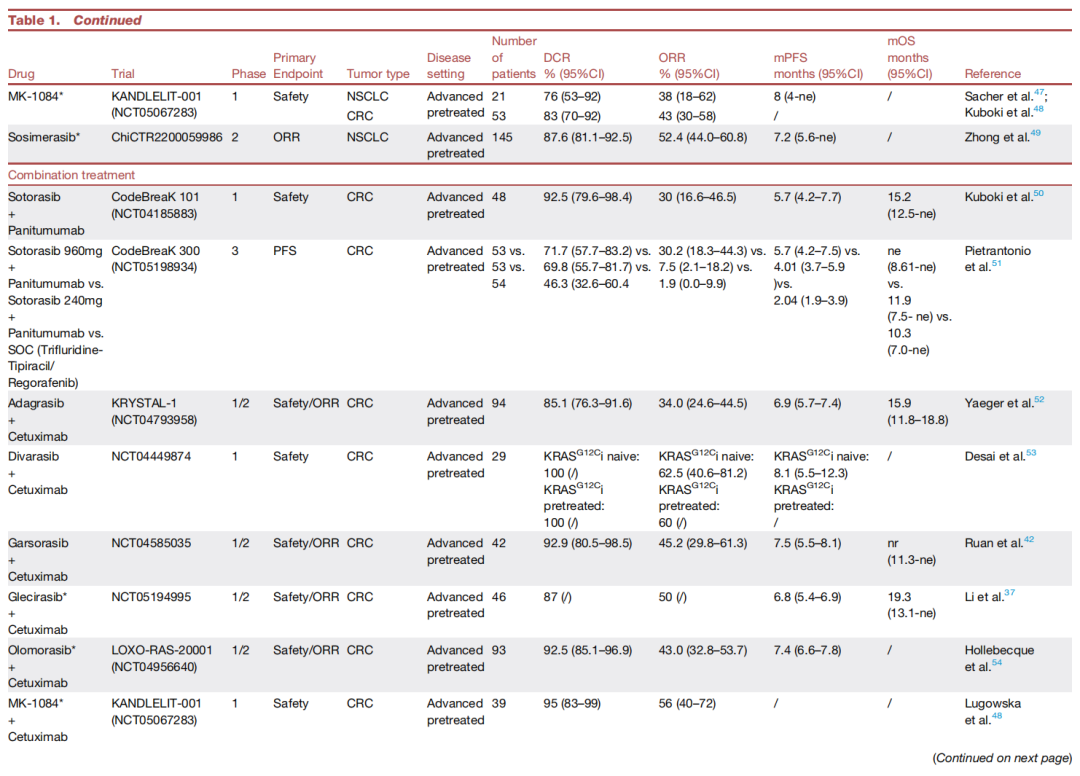
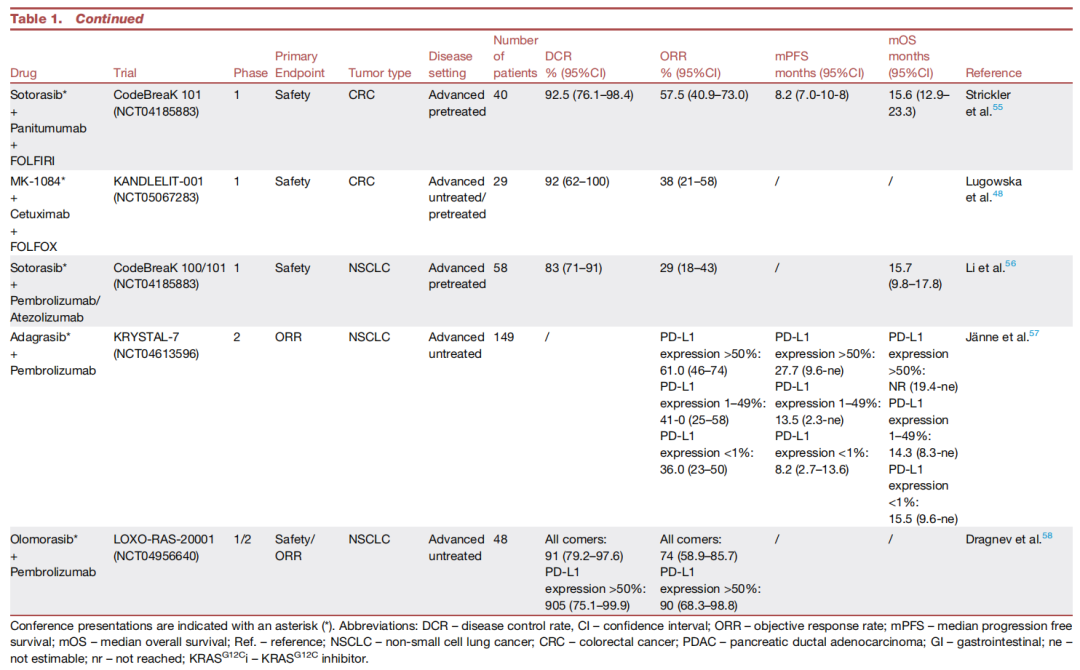
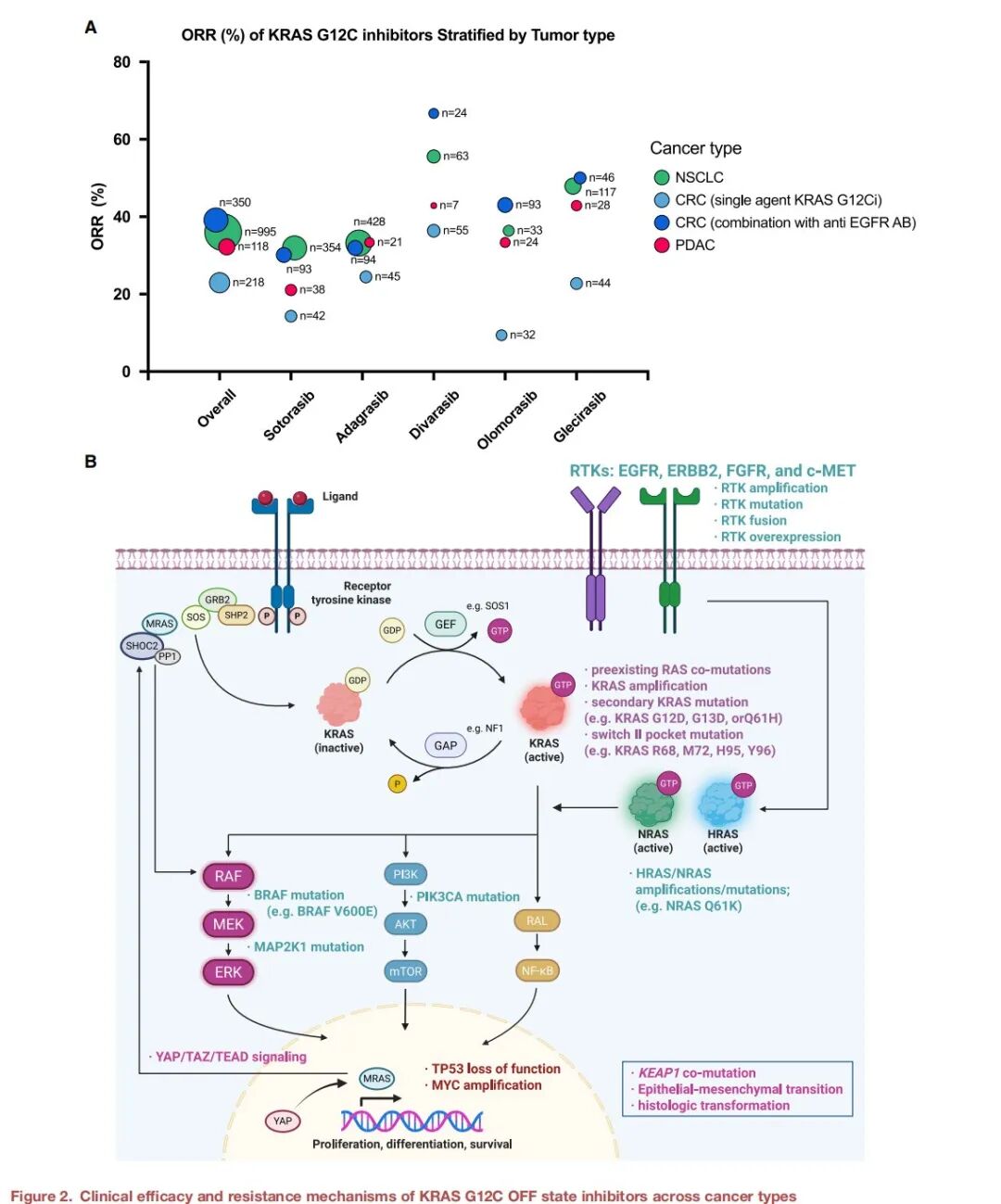
图2. KRAS G12C "关闭"状态抑制剂在不同癌症类型中的临床疗效及耐药机制(A)不同KRAS G12C "关闭"状态抑制剂在不同肿瘤类型中的临床活性。 该气泡图显示了各种KRAS G12C "关闭"状态抑制剂(sotorasib, adagrasib, divarasib, olomorasib, glecirasib)的总缓解率,并按肿瘤类型进行了分层;"总体"代表了所有抑制剂数据的合并结果。图中仅显示在非小细胞肺癌、结直肠癌(KRAS G12C抑制剂单药治疗)、结直肠癌(与抗EGFR抗体联合治疗)和胰腺导管腺癌这四种适应症中均有ORR数据的抑制剂。分析排除了先前接受过KRAS G12C抑制剂治疗的患者。气泡大小对应患者数量。(B)对KRAS G12C "关闭"状态抑制剂的耐药机制。 该示意图概述了介导对KRAS G12C "关闭"状态抑制剂内在或获得性耐药的信号通路和遗传改变。上游受体酪氨酸激酶(如EGFR, ERBB2, FGFR, c-MET)通过扩增、突变、融合或过表达而激活,可恢复RAS通路信号传导。靶上机制包括KRAS扩增、二次突变(如G12D, G13D, Q61H)或开关II口袋突变(如R68, M72, H95, Y96)。耐药也可能源于其他RAS亚型的改变,包括HRAS/NRAS扩增或激活突变(如NRAS Q61K)。下游改变包括BRAF(如V600E)、MAP2K1或PIK3CA的激活突变,以及TP53缺失、MYC扩增或KEAP1共突变。非遗传机制包括上皮-间质转化、组织学转化以及旁路通路(如YAP/TAZ/TEAD信号)的激活。
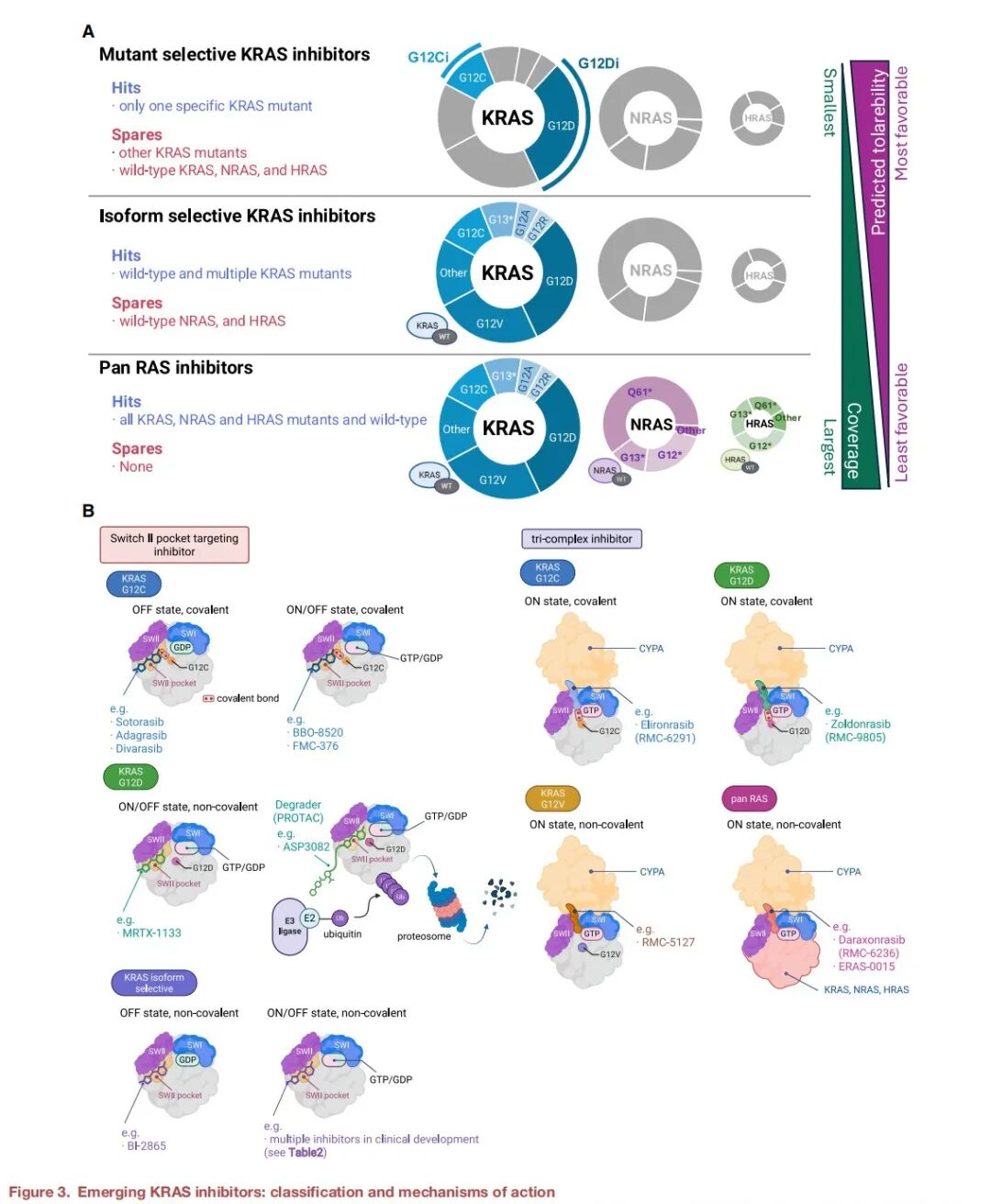
图3. 新兴KRAS抑制剂的分类与作用机制.(A)KRAS抑制剂的分类及其活性谱(突变选择性 vs. 亚型选择性 vs. 泛RAS)。 示意图根据靶点选择性对RAS抑制剂进行分类。突变选择性抑制剂设计用于靶向单一的KRAS突变体(如KRAS G12C或G12D),而不影响其他KRAS突变体以及野生型的KRAS、NRAS和HRAS。亚型选择性抑制剂将其活性扩展至多种KRAS突变体以及野生型KRAS,但不影响NRAS和HRAS。相比之下,泛RAS抑制剂对所有RAS亚型(KRAS, NRAS, HRAS)的突变体和野生型蛋白均起作用。其活性谱范围从最窄(突变选择性)到最宽(泛RAS),这对治疗效能、预测的耐受性及潜在的靶点毒性具有不同影响。(B)新型RAS抑制剂的作用机制(靶向开关II口袋的抑制剂 vs. 三元复合物抑制剂)。 该示意图概述了新型RAS抑制剂,重点说明了其结合模式、作用机制、构象状态选择性和靶点特异性的主要差异。开关II口袋抑制剂包括KRASG12C的共价"关闭"状态抑制剂(如sotorasib, adagrasib, divarasib),以及新兴的能够同时结合"开启"和"关闭"状态KRAS ^G12C的共价药物(如BBO-8520, FMC-376)。对于KRASG12D,开关II口袋抑制剂包括"开启/关闭"状态抑制剂(如MRTX-1133)和降解剂(如ASP3082)。也开发出了KRAS亚型选择性开关II口袋抑制剂,有的仅靶向"关闭"状态(如BI-2865),有的则同时靶向"开启"和"关闭"状态(多种在研药物)。相比之下,三元复合物抑制剂通过与亲环蛋白A形成三元复合物来靶向KRAS的"开启"状态。例子包括elironrasib(RMC-6291,共价G12C选择性)、zoldonrasib(RMC-9805,共价G12D选择性)、RMC-5127(非共价G12V选择性),以及作为泛RAS抑制剂的daraxonrasib(RMC-6236)和ERAS-0015。
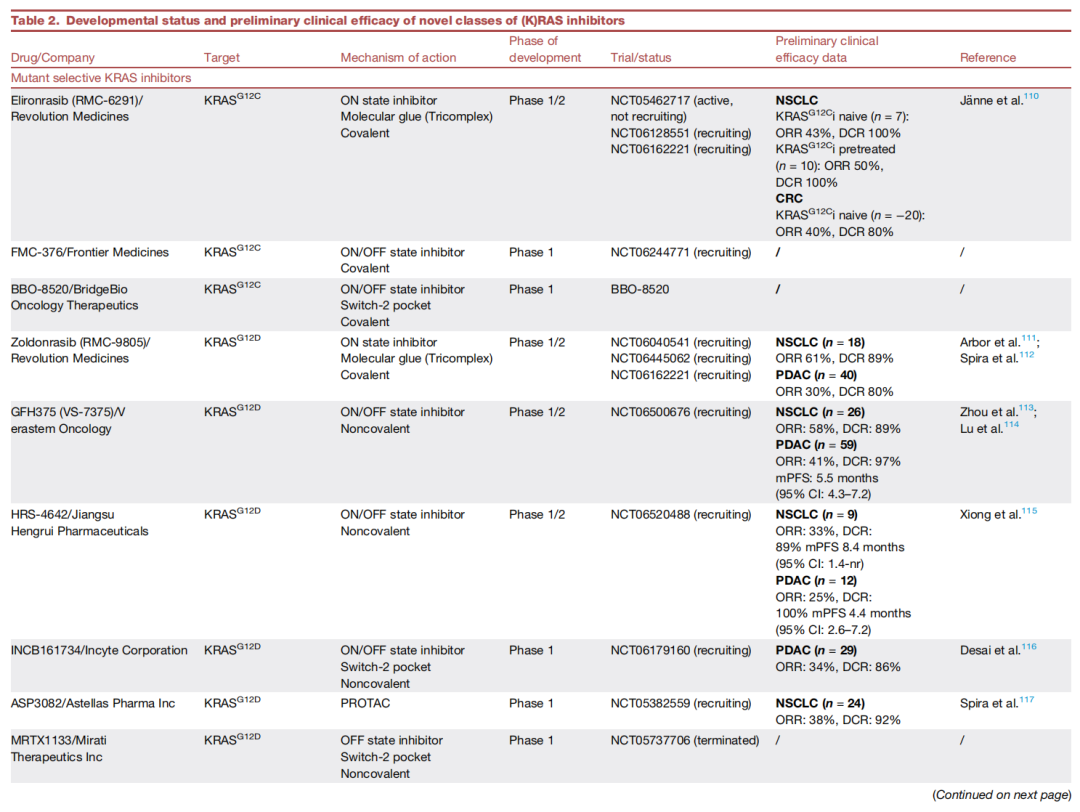
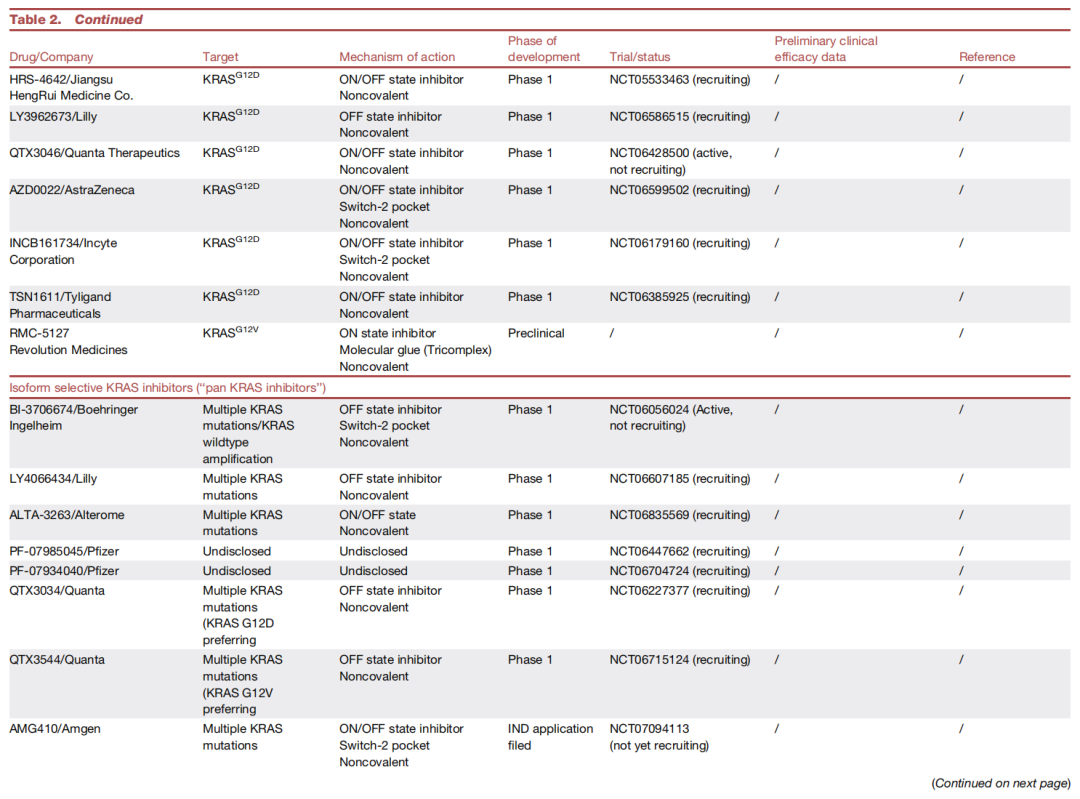
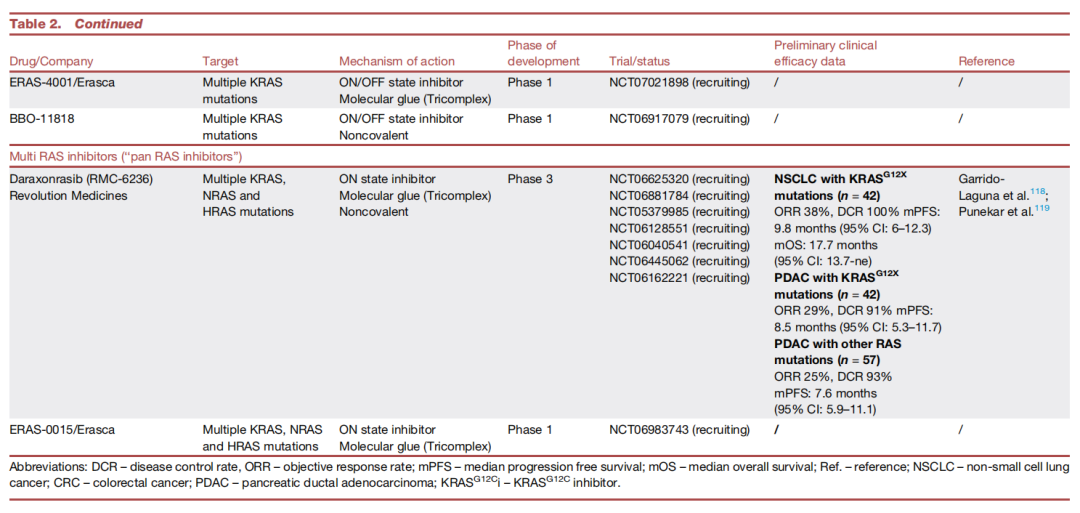
结论
突变选择性KRAS G12C抑制剂Sotorasib(索托拉西布)和Adagrasib(阿达格拉西布)的临床开发和批准证实了,直接药理靶向RAS(长期以来被认为是不可成药的)在临床上是可行的。然而,这些药物局限性凸显了需要更有效的方法来克服耐药性。新兴类别的(K)RAS抑制剂具有更强的效力、更广的靶点覆盖范围和不同的作用机制,有望将益处扩展到更广泛的RAS驱动癌症谱系。实现这一愿景需要更深入地了解耐药机制、指导患者选择的预测性生物标志物,以及利用互补脆弱性的合理药物组合。随着这些进步,有效且持久地抑制RAS这一长期难以实现的目标,可能转化为对全球数百万RAS驱动癌症患者的持久益处。
参考文献:Riedl, Jakob M. et al., Emerging landscape of KRAS inhibitors in cancer treatment, 2026, Cancer Cell, Volume 0, Issue 0
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-02-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号