IDFlow:基于能量驱动幂等流映射的 3D 分子结构生成方法

IDFlow:基于能量驱动幂等流映射的 3D 分子结构生成方法

MindDance
发布于 2026-01-08 13:30:33
发布于 2026-01-08 13:30:33
在结构生物学与药物研发领域,3D分子结构生成始终是核心难题——从蛋白质对接预测到全新蛋白质设计,精准的分子构象建模直接决定了药物效用评估、酶功能优化等关键任务的成败。近年来,生成式模型(如扩散模型、流匹配)虽在该领域取得进展,但如何在保证计算效率的同时,提升模型对分子能量景观的刻画能力,仍是未被完全解决的挑战。
近期,来自瑞典皇家理工学院、斯德哥尔摩大学等机构的团队在arXiv上发表的《Energy-Based Flow Matching for Generating 3D Molecular Structure》一文,提出了一种基于能量视角的流匹配框架(IDFlow),通过将能量最小化原则与流匹配模型深度融合,实现了分子对接与蛋白质骨架生成性能的显著提升。本文将从研究背景、核心创新、方法细节与实验验证四个维度,为大家解析这一工作。

一、研究背景:从“确定性预测”到“概率性生成”的范式转变
传统的分子结构预测方法(如AlphaFold2、RoseTTAFold)多基于回归模型,通过深度神经网络输出单一的“最优”构象。这类方法虽在蛋白质折叠等任务中表现出色,但存在两个关键局限:一是无法捕捉分子固有的“不确定性”——许多分子(如酶与底物复合物)存在多种热力学稳定构象,单一预测无法覆盖全部功能相关态;二是缺乏对分子能量景观的显式建模,生成的构象可能不符合物理化学约束(如键长、范德华力),需额外进行能量松弛优化,增加了计算成本。
为解决上述问题,生成式模型成为新的研究方向。其中,扩散模型(如DiffDock)通过逐步去噪过程建模分子构象分布,能生成多模态构象,但采样过程依赖大量迭代,计算效率较低;流匹配模型(如HarmonicFlow、FrameFlow)则通过学习从源分布(随机构象)到目标分布(真实分子结构)的连续映射,实现了更快的采样,但传统流匹配的目标函数仅关注“轨迹拟合”,未充分利用分子能量这一核心物理先验,导致生成构象的物理有效性与精度仍有提升空间。
该研究的核心洞察在于:分子结构的稳定构象本质上是能量函数的局部极小值(符合玻尔兹曼分布 ),而现有流匹配模型未将这一物理规律显式融入训练过程。因此,团队提出将“能量视角”引入流匹配,通过构建能量驱动的目标函数,让模型在学习映射关系的同时,主动向能量极小值区域收敛——这正是IDFlow的核心设计理念。
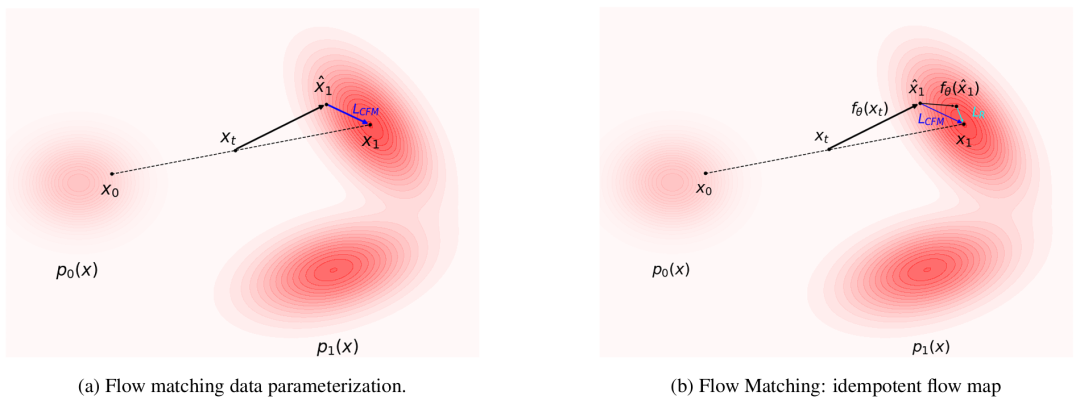
<b>标准流匹配与 IDFlow 的训练范式对比。</b>其中,(a) 标准流匹配直接基于插值样本预测目标构象,仅依赖条件流匹配损失优化;(b) IDFlow 则通过深度神经网络表示的映射函数,对预测构象进行迭代预测与精修,以 “精修后的输入 - 输出结构差异” 作为能量估计,同时结合<b>条件流匹配损失</b>与<b>精修损失</b>优化,体现了其 “生成 - 精修一体化” 的核心设计。
标准流匹配与 IDFlow 的训练范式对比。其中,(a) 标准流匹配直接基于插值样本预测目标构象,仅依赖条件流匹配损失优化;(b) IDFlow 则通过深度神经网络表示的映射函数,对预测构象进行迭代预测与精修,以 “精修后的输入 - 输出结构差异” 作为能量估计,同时结合条件流匹配损失与精修损失优化,体现了其 “生成 - 精修一体化” 的核心设计。
二、核心创新:能量驱动的幂等流映射(Idempotent Flow Map)
IDFlow的创新并非孤立的技术改进,而是围绕“能量建模-流匹配优化-迭代精修”形成的完整框架,其中两个核心概念贯穿始终:能量函数的显式构建与幂等流映射的学习。
1. 能量函数:从“轨迹拟合”到“能量极小化”的目标重构
传统流匹配(如Conditional Flow Matching, CFM)的目标函数仅关注“预测构象与真实构象的L2误差”,即 ()(其中 () 为流映射,() 为真实构象)。这种目标仅能保证“轨迹上的点向真实构象靠近”,但无法确保生成的构象处于能量极小值区域——部分预测构象可能虽与真实构象误差较小,却因违反物理约束(如原子间距过近)而不具备实际意义。
IDFlow则将能量函数显式引入目标函数。团队提出:以“生成构象的自重构误差”作为能量度量,即 (),其中 () 是流模型生成的构象,(G) 是一个“精修函数”。直观上,若生成构象 () 处于能量极小值区域(物理有效),则其自重构误差会很小;反之,若构象违反物理约束,自重构误差会显著增大。
通过将这一能量函数融入训练目标,IDFlow实现了两个关键突破:一是让模型在学习流映射时,同时向“低能量区域”收敛,无需额外的能量松弛步骤;二是通过“对比采样”(从流模型中采样多个 ()),进一步塑造能量景观——让“好构象”(低能量)与“坏构象”(高能量)形成明确的能量差,提升模型对构象质量的区分能力。
2. 幂等流映射:让流模型兼具“生成”与“精修”双重功能
在传统流匹配中,流映射 () 的作用是“一次性预测”——从源分布的随机构象直接映射到目标构象。而IDFlow通过能量函数的约束,意外发现:流映射可以被训练成幂等函数(Idempotent Function),即满足 ()。
这一性质的意义在于:流模型不再是“一次性生成器”,而是兼具“生成”与“精修”的双重功能。具体来说,当模型生成初始构象 () 后,再次输入同一流映射 () 时,构象不会发生显著变化(幂等性)——这意味着生成的构象已处于能量极小值区域;若初始构象未达稳定态,多次迭代映射会逐步将其“精修”至低能量区域。这种设计与AlphaFold2的“迭代精修”机制异曲同工,但IDFlow将其原生融入流匹配的训练过程,无需额外模块。
从理论上看,幂等流映射的收敛性可通过能量函数保证:当迭代次数足够多时,(),即最终构象必然是能量函数的极小值点——这为生成构象的物理有效性提供了理论保障。
三、方法细节:训练与推理的完整流程
IDFlow的方法设计围绕“如何高效学习幂等流映射”展开,其训练与推理流程既保留了流匹配的高效性,又融入了能量驱动的精修逻辑,具体可分为三个关键步骤:
1. 数据参数化与流匹配目标融合
IDFlow采用流匹配的“(x_1)参数化”策略——即流映射 (f_{\theta,t}) 直接预测目标构象 (),而非中间向量场。这种参数化方式的优势在于:能直接与能量函数的L2形式对齐,简化目标函数的计算。
训练过程中,模型采用“双阶段目标”:
- • 50%的训练时间用于传统CFM目标训练,确保流映射具备从源分布到目标分布的基础映射能力;
- • 剩余50%的训练时间用于幂等目标训练:先通过流映射生成 ()(并 detach 梯度,避免干扰基础映射学习),再以 () 为损失,训练流映射的幂等性。
这种设计既保证了流匹配的高效采样特性,又通过幂等目标赋予模型精修能力,且仅引入一个额外超参数 ()(最大迭代精修次数),计算开销与传统流匹配基本相当。
2. 黎曼流匹配的扩展:适配蛋白质骨架的流形特性
分子结构生成中,蛋白质骨架的建模需考虑其特殊的几何约束——蛋白质残基的构象本质上处于SE(3)流形(旋转+平移的组合空间),而非欧几里得空间。传统流匹配在欧几里得空间中表现良好,但直接应用于流形时会产生几何畸变。
IDFlow通过“黎曼流匹配”(Riemannian Flow Matching)解决这一问题:在SE(3)流形上定义测地线(Geodesic)作为构象的插值路径,而非欧几里得空间的线性插值。具体来说,对于蛋白质残基的旋转部分 (),采用指数映射与对数映射计算测地线插值:(其中 和 操作通过罗德里格斯公式高效计算);对于平移部分 ,则仍采用线性插值 。
这种扩展确保了IDFlow在蛋白质骨架生成任务中,能严格遵循SE(3)流形的几何约束,生成的蛋白质构象在旋转、平移变换下保持物理一致性——这也是其在蛋白质骨架生成任务中表现优异的关键原因之一。
3. 预测-精修采样器:高效的推理过程
IDFlow的推理过程被设计为“预测-精修采样器”(Predictor-Refiner Sampler),流程如下:
- 1. 从源分布 采样初始随机构象 ;
- 2. 设定采样步数 与步长 ,初始化当前构象 ;
- 3. 对每个时间步 :
- • 调用流映射 预测目标构象 ;
- • 再次调用 对 进行精修(利用幂等性);
- • 按欧拉法更新当前构象:;
- 4. 完成所有步数后,输出最终构象 。
这种采样过程的优势在于:每一步都包含“预测”与“精修”,无需额外的后处理步骤;同时,由于幂等性的存在,即使采样步数较少(如10步),也能生成高质量构象——实验表明,IDFlow在仅使用 10步采样(20次函数评估) 时,性能仍优于传统流匹配的20步采样结果。
四、实验验证:多任务、多数据集的全面超越
为验证IDFlow的有效性,团队在分子对接(单配体、多配体)与蛋白质骨架生成两大核心任务上,基于4个公开数据集(PDBBind v2020、Binding MOAD、SCOPe、PDB子集)进行了系统性实验,并与当前主流方法(如HarmonicFlow、DiffDock、FrameFlow)进行对比。
1. 分子对接任务:精度与模式覆盖能力双提升
分子对接的核心指标是“生成构象与真实构象的RMSD误差”——RMSD<2Å表示构象高度准确,RMSD<5Å表示构象具备功能相关性。
- • 单配体对接(PDBBind数据集):在序列相似性拆分与时间拆分的两种测试场景下,IDFlow均显著优于基线方法。例如,在序列相似性拆分的“距离口袋”任务中,IDFlow的RMSD<2Å比例达到35.6%,较HarmonicFlow(30.1%)提升5.5个百分点;在时间拆分的“半径口袋”任务中,IDFlow的RMSD<2Å比例达到34.7%,较HarmonicFlow(28.3%)提升6.4个百分点。
- • 多配体对接(Binding MOAD数据集):多配体对接需模型学习同一蛋白质与多个配体的结合模式,难度更高。IDFlow在RMSD<5Å指标上达到83.1%,较HarmonicFlow(75.0%)提升8.1个百分点,表明其对多模态构象的覆盖能力更强。
此外,Top-k精度实验(评估模型从k个生成构象中选出高质量构象的能力)显示,IDFlow在k=5、10、40的场景下,均能稳定优于基线——例如,在k=5时,IDFlow的RMSD<2Å比例达到52.0%,较HarmonicFlow(47.5%)提升4.5个百分点,证明其生成构象的“整体质量”更优。
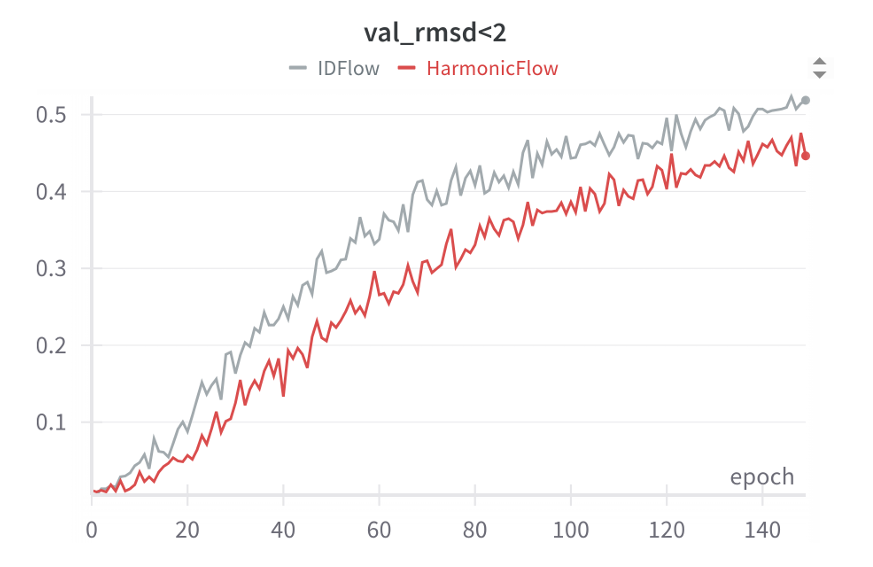
<b>IDFlow 与 HarmonicFlow 在分子对接任务中的验证指标曲线。</b>对比了 IDFlow 与 HarmonicFlow 在 PDBBind 数据集 “半径口袋 - 时间拆分” 场景下的训练收敛过程。曲线显示,IDFlow 的收敛速度显著快于 HarmonicFlow—— 在训练早期(约 20 epoch)即达到 HarmonicFlow 后期(约 100 epoch)的性能水平;且 IDFlow 最终的验证集 RMSD<2Å 比例(约 0.45)远高于 HarmonicFlow(约 0.3)。
IDFlow 与 HarmonicFlow 在分子对接任务中的验证指标曲线。对比了 IDFlow 与 HarmonicFlow 在 PDBBind 数据集 “半径口袋 - 时间拆分” 场景下的训练收敛过程。曲线显示,IDFlow 的收敛速度显著快于 HarmonicFlow—— 在训练早期(约 20 epoch)即达到 HarmonicFlow 后期(约 100 epoch)的性能水平;且 IDFlow 最终的验证集 RMSD<2Å 比例(约 0.45)远高于 HarmonicFlow(约 0.3)。
2. 蛋白质骨架生成:设计性与结构多样性兼顾
蛋白质骨架生成的核心指标包括“设计性”(生成构象能否通过逆折叠得到功能序列)、“多样性”(生成构象间的结构差异)与“新颖性”(生成构象与天然蛋白质的差异)。
- • PDB子集实验(蛋白质长度100-300):IDFlow在设计性指标上表现突出——当使用100次函数评估(NFEs)时,设计性达到87.1%,较FrameFlow(82.4%)提升4.7个百分点;当使用200次NFEs时,设计性进一步提升至92.7%,接近依赖序列信息的FoldFlow2(94%),但IDFlow无需序列输入,通用性更强。
- • 结构特性:与FoldFlow2易生成单一螺旋结构不同,IDFlow生成的构象同时包含α-螺旋与β-折叠,更符合天然蛋白质的结构多样性;此外,IDFlow的多样性与新颖性指标与基线相当,证明其在提升设计性的同时,未牺牲结构的多样性与创新性。
- • 效率优势:IDFlow生成长度为100的蛋白质骨架仅需3.3秒,较FrameFlow(6.6秒)效率提升一倍,这得益于其幂等流映射的高效精修机制——无需额外迭代即可生成稳定构象。
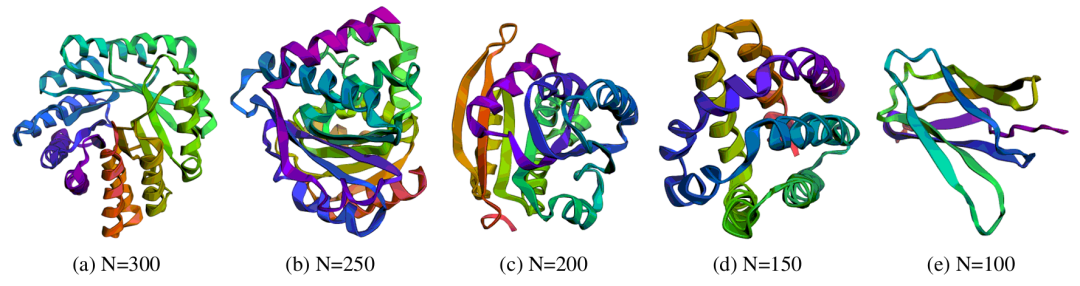
<b>IDFlow 生成的不同长度可设计蛋白质骨架。</b>该图展示了 IDFlow 在蛋白质骨架生成任务中,针对不同长度(N=100,150,200,250,300)蛋白质生成的可设计构象,对应子图 (a) 至 (e)。生成的蛋白质骨架采用非冗余拓扑结构,且同时包含 α- 螺旋与 β- 折叠两种关键二级结构,与天然蛋白质的结构多样性特征相符。这一结果直接佐证了 IDFlow 的优势 —— 区别于 FoldFlow2 等基线模型易 “结构坍塌”(仅生成螺旋结构)的问题,IDFlow 能生成符合天然蛋白质结构规律的多样化构象,且所有展示构象均通过 “设计性” 验证(可通过逆折叠得到功能序列),证明其具备实际蛋白质工程应用潜力。
IDFlow 生成的不同长度可设计蛋白质骨架。该图展示了 IDFlow 在蛋白质骨架生成任务中,针对不同长度(N=100,150,200,250,300)蛋白质生成的可设计构象,对应子图 (a) 至 (e)。生成的蛋白质骨架采用非冗余拓扑结构,且同时包含 α- 螺旋与 β- 折叠两种关键二级结构,与天然蛋白质的结构多样性特征相符。这一结果直接佐证了 IDFlow 的优势 —— 区别于 FoldFlow2 等基线模型易 “结构坍塌”(仅生成螺旋结构)的问题,IDFlow 能生成符合天然蛋白质结构规律的多样化构象,且所有展示构象均通过 “设计性” 验证(可通过逆折叠得到功能序列),证明其具备实际蛋白质工程应用潜力。
五、总结与展望:能量视角为分子生成开辟新路径
IDFlow的核心贡献在于:将分子结构的“能量本质”与流匹配的“高效映射”深度融合,通过幂等流映射这一简洁设计,同时解决了传统生成模型“物理有效性不足”与“采样效率低”两大痛点。其创新点可概括为三点:
- 1. 能量驱动的目标重构:将自重构误差作为能量函数,让模型原生学习物理约束,无需额外能量松弛;
- 2. 幂等流映射的学习:让流模型兼具生成与精修功能,简化模型架构并提升构象质量;
- 3. 黎曼流匹配的扩展:适配蛋白质骨架的SE(3)流形特性,确保几何一致性。
从应用角度看,IDFlow为药物研发与蛋白质工程提供了新工具:在药物对接中,其高精度构象可减少实验筛选的盲目性;在蛋白质设计中,其高设计性构象为酶改造、抗体设计等任务提供了高质量候选结构。
未来,该团队计划进一步融合“生物物理启发的能量函数”(如Amber力场),让模型更精准地捕捉分子间的非键相互作用;同时,探索IDFlow在分子动力学模拟、蛋白质-蛋白质相互作用预测等更复杂任务中的应用——可以预见,能量视角的流匹配方法将成为分子生成领域的重要研究方向。
对相关领域研究者而言,这篇论文不仅提供了一种高性能的分子生成方法,更重要的是展示了“物理先验与生成模型融合”的研究思路——这种思路为解决其他复杂系统的生成问题(如材料设计、细胞结构建模)提供了宝贵借鉴。
参考文献
论文:
Energy-Based Flow Matching for Generating 3D Molecular Structure 代码:
https://github.com/CaviarLover/IDFlow
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-28,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号