J. Chem. Inf. Model. | Qsarna:智能化学空间导航工具在药物设计中的应用

J. Chem. Inf. Model. | Qsarna:智能化学空间导航工具在药物设计中的应用

MindDance
发布于 2026-01-08 13:05:11
发布于 2026-01-08 13:05:11
Qsarna: An Online Tool for Smart Chemical Space Navigation in Drug Design
期刊: Journal of Chemical Information and Modeling 链接: https://doi.org/10.1021/acs.jcim.5c00720 平台: https://qsarna.com/ 简介: 本文介绍了 Qsarna,一款综合性在线平台,其创新点在于将机器学习的活性预测与传统分子对接相结合,同时融入片段基生成模型,实现智能化学空间导航以优化药物设计流程。该平台通过整合分子对接、QSAR 机器学习模型和片段基生成设计三种工具,形成正交筛选机制,并提供用户友好的界面和数据管理功能。在实验中,研究团队利用 ChEMBL 数据库中的化合物训练模型,针对单胺氧化酶 B(MAO-B)进行虚拟筛选,从 MolPort 库中筛选并实验验证出 3 个具有纳摩尔级效力的新抑制剂,其中最有效的化合物 IC50 达 1.37 nM。该论文表明,Qsarna 通过整合多种计算方法并降低使用门槛,为药物发现提供了高效且易于获取的工具,有助于加速药物研发进程。

药物发现领域长期面临着研发周期冗长、资源投入巨大的核心挑战,据统计,每款获批药物的研发成本已超过25亿美元,且研发周期常跨越10至15年。这种高投入与临床试验中的高淘汰率,促使业界不断探索更高效的研发范式。计算方法的引入为解决这一困境提供了新思路,其中虚拟筛选技术通过对海量化合物的系统性评估,显著减少了需实验验证的候选分子数量,从而缩短研发周期并降低成本。然而,现有工具要么受限于商业授权模式,要么存在功能碎片化问题,难以满足整合化、低成本的研究需求。在此背景下,Qsarna作为一款创新性在线平台的出现,为药物设计领域的智能化学空间导航提供了全新解决方案。
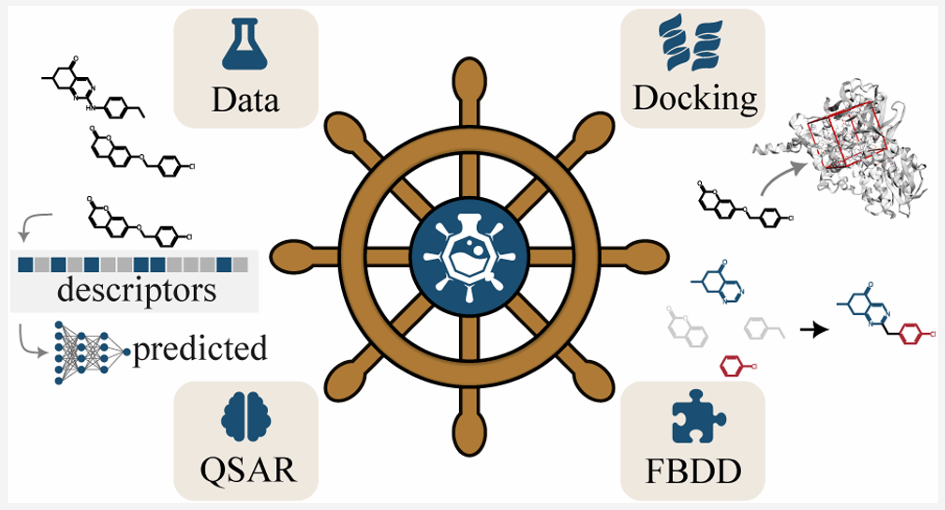
平台核心架构与技术整合
Qsarna的核心优势在于其对多种虚拟筛选技术的深度整合,构建了一套涵盖分子对接、机器学习预测及片段生成设计的完整工作流。在分子对接模块,平台采用基于AutoDock Vina开发的Smina软件作为核心引擎,支持用户自定义对接参数(包括构象搜索详尽度、生成构象数量及互变异构体数量等),并通过实时可视化界面准确定义结合口袋区域。值得注意的是,该模块实现了配体预处理的全自动化,包括质子化状态计算(基于OpenBabel)、立体异构体生成及低能构象优化(基于RDKit),确保了对接结果的可靠性与一致性。通过DUD-E数据库的诱饵化合物测试显示,在三个分子靶点中的两个上,Qsarna的性能可与商业软件Glide相媲美。
配体基于的活性预测模块是Qsarna智能化的关键体现。平台整合AutoML工具链,可自动构建随机森林、支持向量机及人工神经网络三种模型,采用Morgan指纹、Avalon指纹、MACCS键及RDKit分子描述符作为特征输入。模型训练过程支持数据自动划分(训练集、验证集、测试集)与超参数优化,可处理连续活性值(如IC50)或二元分类(基于阈值定义活性类别)任务。特别值得一提的是,该模块支持基于对接结果训练QSAR模型,通过这种方式可有效提高对接分子的优先级排序,减少假阳性结果并挖掘非直观的构效关系。同步集成的ADMET预测器涵盖血脑屏障通透性、hERG结合、CACO-2通透性等关键属性,其性能经与ADMETlab、admetSAR等主流工具对比验证,达到行业先进水平。
片段基于的生成设计模块则拓展了化学空间探索的边界。该模块基于CRET算法,通过已知连接子将具有结合能力的片段进行系统性连接与扩展,生成的分子既保留关键相互作用,又确保合成可行性。生成过程严格遵循片段在结合口袋中的空间位置约束,后续可通过平台内置对接模块对新分子进行重新对接,优先保留片段初始位置附近的构象。这一功能对于发现新型化学型、规避专利结构具有重要价值,基准测试表明其能生成独特且多样的可合成化合物库。
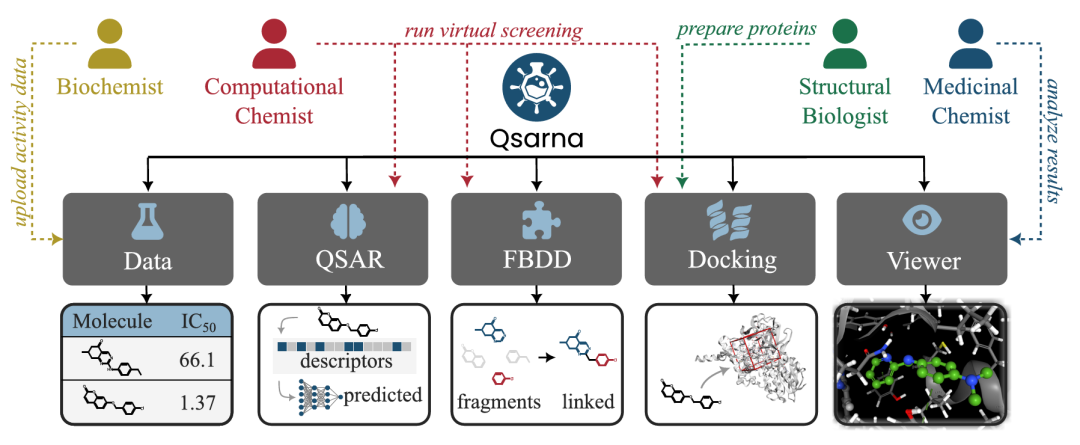
该图展示了 Qsarna 的核心功能模块,呈现项目仪表板的布局。平台支持用户创建协作项目,数据模块可上传化合物结构和活性测量结果;虚拟筛选模块通过 QSAR 模型和分子对接(针对上传化合物或自动关联片段)识别潜在分子;结果可视化模块能展示配体 - 靶点相互作用,体现了工具整合多步骤流程的特点。
该图展示了 Qsarna 的核心功能模块,呈现项目仪表板的布局。平台支持用户创建协作项目,数据模块可上传化合物结构和活性测量结果;虚拟筛选模块通过 QSAR 模型和分子对接(针对上传化合物或自动关联片段)识别潜在分子;结果可视化模块能展示配体 - 靶点相互作用,体现了工具整合多步骤流程的特点。
系统设计与用户体验优化
Qsarna采用Django框架(Python 3)开发,后端使用PostgreSQL数据库存储用户数据(化合物库、分析结果等),非结构化分子数据(对接构象、蛋白质结构)以SDF和PDB格式保存,机器学习模型则采用二进制文件存储。计算密集型任务通过Celery队列管理,用户可实时查看任务状态。部署方式支持本地Docker容器与AWS云架构两种模式,云部署方案利用Amazon RDS确保数据完整性与可用性,通过EC2实例运行主应用,结合SQS与Batch服务实现计算任务的动态分配与资源弹性扩展。
用户体验设计上,平台采用直观的图形界面,提供交互式入门教程与详细PDF手册,新用户注册后会自动获得示例项目,无需上传数据即可熟悉功能。项目管理支持多用户协作,所有操作通过项目仪表板的网格 tile 布局实现,包括化合物库上传、筛选结果分析及实验设计等功能。数据管理机制确保ACID合规性,实现化合物自动去重与记录合并,支持库共享与评论功能,为协作研究提供便利。
分子可视化模块增强了结果解读的直观性。对接过程中实时显示边界框,筛选结果可通过内置查看器直接展示,自动标注配体-蛋白相互作用及邻近氨基酸。用户可对构象进行5分制评分,结合分子动力学可视化模块,还能分析配体相互作用随时间的变化趋势。

登陆之后的主页图。可以看到Monoamine Oxidase B项目已经预定义好了。
登陆之后的主页图。可以看到Monoamine Oxidase B项目已经预定义好了。
实证研究:单胺氧化酶抑制剂的发现
为验证平台效能,研究团队采用Qsarna开展单胺氧化酶(MAO)抑制剂的虚拟筛选。首先上传ChEMBL数据库中所有含MAO-A/B的Ki或IC50值的化合物,经平台自动预处理(去重、盐离子去除)后,将问题转换为二元分类任务(活性阈值设为100 nM),分别训练MAO-A和MAO-B模型,ROC AUC值分别达到0.92和0.88。
利用 top 3 模型筛选MolPort化合物库,经0.65分类阈值过滤得到556个化合物,进一步对接至MAO-A(PDB:2BXR)和MAO-B(PDB:2V5Z)结构,结合结合构象可视化检查与经济性考量,最终选取19个化合物进行实验验证。结果显示,通过Merck抑制剂筛选试剂盒检测,发现3个新型MAO-B抑制剂,其中最优化合物的IC50达到1.37 nM, potency显著优于先前研究报道。该案例充分证明,Qsarna通过多方法整合(机器学习筛选→分子对接验证→实验确认),可有效提高 hit 发现效率,为复杂靶点的药物设计提供有力支持。
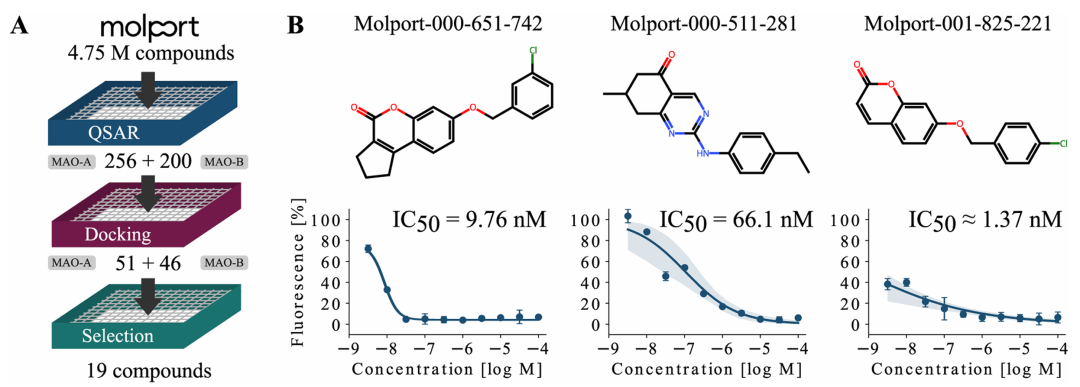
MAO 抑制剂案例研究结果。A 部分显示对大型 MolPort 库的筛选流程:先通过基于 ChEMBL 数据训练的 QSAR 模型筛选,再经分子对接和视觉检查进一步过滤;B 部分呈现 3 个最有效的 MAO-B 抑制剂在生化分析中的结果,包括它们的化学标识及剂量 - 反应曲线,其中最 potent 化合物的 IC50 约为 1.37 nM,验证了平台的筛选效能。
MAO 抑制剂案例研究结果。A 部分显示对大型 MolPort 库的筛选流程:先通过基于 ChEMBL 数据训练的 QSAR 模型筛选,再经分子对接和视觉检查进一步过滤;B 部分呈现 3 个最有效的 MAO-B 抑制剂在生化分析中的结果,包括它们的化学标识及剂量 - 反应曲线,其中最 potent 化合物的 IC50 约为 1.37 nM,验证了平台的筛选效能。
结语与展望
Qsarna的推出填补了免费、综合型虚拟筛选平台的空白,其核心价值在于:通过整合结构基于与配体基于方法,克服单一技术的局限性;通过自动化工作流与用户友好设计,降低计算药物设计的技术门槛;通过免费向学术界开放,促进先进方法的普及应用。该平台特别适合实验数据有限的靶点研究、hit 扩展及减少化学偏差等场景,有望加速药物发现进程。
目前Qsarna已在线开放(https://qsarna.com),用户每月享有一定的计算额度,学术项目可申请提升限额。作为一款不断进化的工具,其未来发展将聚焦于模型性能优化、更多靶点类型的适配及与实验数据的闭环整合,为药物研发社区提供更强大的技术支撑。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-08-12,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号