Chem. Sci. | 面向计算药物设计的深度学习分子生成:靶蛋白结构的整合与应用

Chem. Sci. | 面向计算药物设计的深度学习分子生成:靶蛋白结构的整合与应用

DrugAI
发布于 2026-01-06 13:17:17
发布于 2026-01-06 13:17:17
DRUGONE
传统药物研发成本高昂且效率低下,候选化合物常因疗效不足或非特异性结合而失败。基于结构的药物设计(SBDD)通过在分子设计中直接整合蛋白靶点结构信息,为提高研发成功率提供了重要途径。本文综述了近年来将蛋白结构引入深度学习分子生成中的方法,从早期基于形状的建模到能够共同预测蛋白–配体结构的协同折叠模型。研究人员重点分析了如何利用三维结构信息生成兼具结合能力与化学合理性的分子,并讨论了该领域未来的发展方向,包括模型可解释性、通用化及物理约束的进一步融合。

药物研发平均成本超过20亿美元,主要瓶颈来自临床阶段的高失败率。约六成候选药物在II–III期临床中因疗效不足被终止,另一重要原因是安全性问题,通常源于非靶点结合。由于临床阴性数据的公开极少,研究人员难以系统学习失败经验,因此提高早期候选分子质量成为关键策略。
药物设计分为两大类:
- 基于配体的药物设计(LBDD):依赖于已知配体结构推测靶点结合特征;
- 基于结构的药物设计(SBDD):直接利用蛋白靶点的三维结构设计新分子。
相比之下,SBDD 类似于“获得锁的蓝图来制作钥匙”,能更直接、精准地构建新分子。然而,其难点在于如何从完整蛋白结构中提取对结合最关键的几何与化学特征,并以机器学习的方式进行高效编码。
结构信息在药物设计中的价值
候选分子失败的主要原因包括结合力不足、选择性差及毒性高。SBDD 通过结合三维结构数据指导分子优化,可同时改善这些方面。理想的生成模型应能在保留化学可行性的同时学习到蛋白–配体相互作用规律,从而设计出高亲和力、低脱靶风险的候选分子。
因此,SBDD 的挑战在于:
- 如何以机器学习模型精确表示蛋白–配体结构特征;
- 如何建立统一、严谨的评价体系,衡量生成分子的新颖性与特异性。
深度学习在结构基础分子设计中的实践考量
靶标结构信息的引入
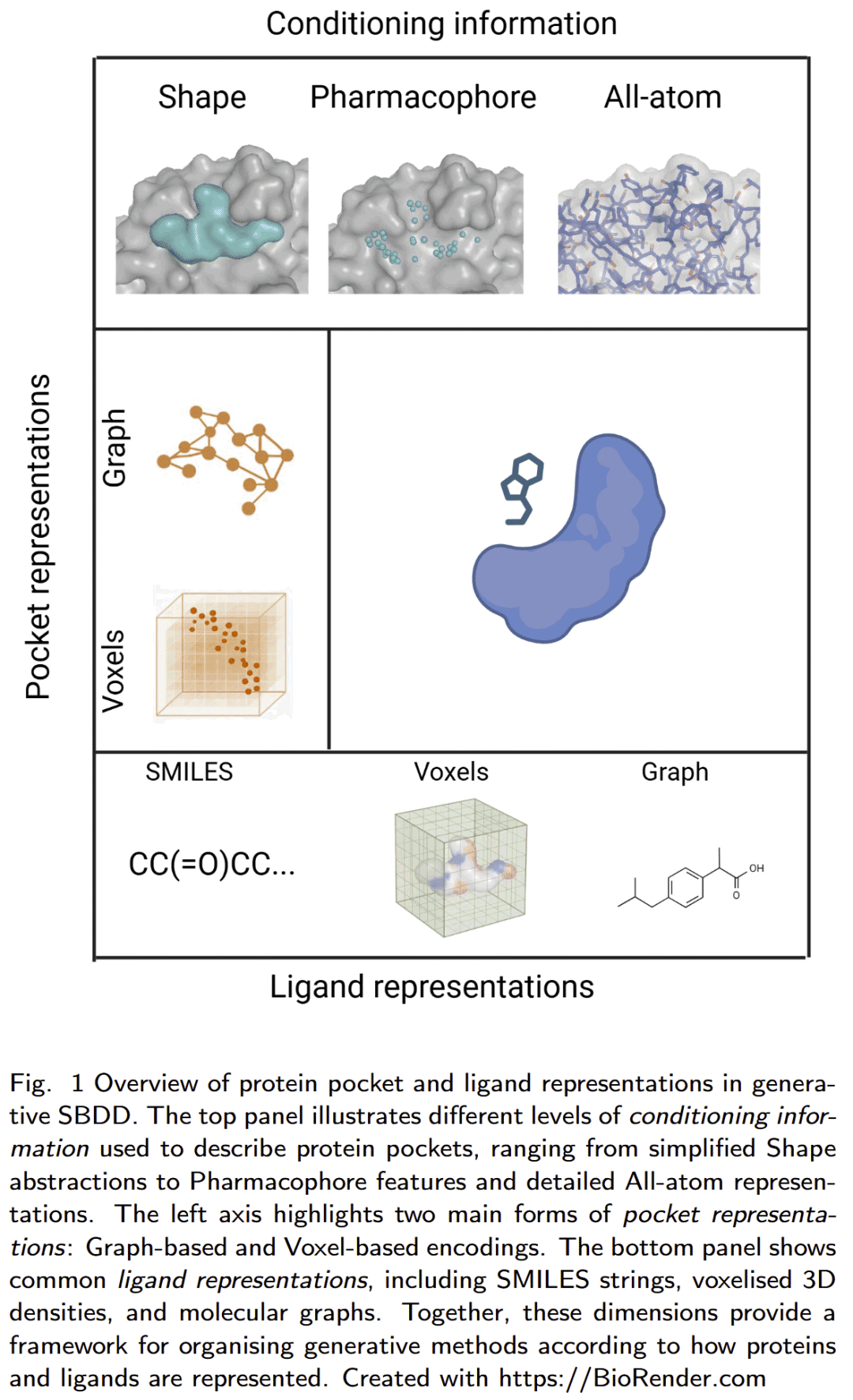
早期方法以形状或药效团映射的方式简化蛋白结合位点表示,例如:
- DESERT:将结合腔体离散化为体素网格;
- DEVELOP、STRIFE:利用药效团点引导图结构生成。
这些方法计算效率高,但无法捕捉所有原子层面的相互作用。随后,全原子图模型成为主流,如 Pocket2Mol 等利用 E(3)-等变图神经网络在三维空间直接生成分子结构,保持旋转平移不变性。
然而,最新基准测试(PoseCheck)表明,即使是先进模型也难以再现真实氢键网络,因此混合方法如 MolSnapper、DiffSBDD 被提出,结合药效团约束与全原子生成以提高生物物理合理性。
数据集与训练挑战
SBDD 模型依赖高质量三维数据。主要来源包括:
- 实验结构库(PDB、PDBbind、Binding MOAD、BioLiP);
- 计算生成库(CrossDocked、DUD-E、AlphaFold DB);
- 混合资源(MISATO、PLINDER);
- 配体预训练集(ZINC、ChEMBL、MOSES、QM9 等)。
实验数据稀缺且偏倚严重,易导致模型过拟合。为此,研究人员普遍采用“预训练 + 微调”策略:先在大规模配体数据上学习化学规律,再在特定蛋白–配体集合上细化训练。
评价指标
评价标准分为两类:
- 分子质量指标:包括有效性(Validity)、药物相似性(QED)、合成可行性(SA)、Lipinski 法则、结构多样性与新颖性;
- 构象与结合质量指标:主要依赖对接评分(Vina/Smina),但存在偏向大分子的风险。
研究人员强调应使用局部能量最小化代替“再对接(redocking)”,以避免掩盖模型生成缺陷。此外,综合指标如形状–颜色相似度与PoseBusters 物理合理性检查被逐渐采纳。

主要技术进展
基于体素网格的生成方法
这类方法通过将结合腔体离散化为三维网格表示蛋白环境:
- DESERT:基于形状条件的自编码器,生成在结合腔体内拟合良好的分子;
- DEVELOP:利用3D药效团约束与CNN–GNN联合架构生成分子连接体;
- STRIFE:融合片段热点图,实现完全结构驱动的片段扩展;
- LiGAN:首个基于全原子网格的生成模型,采用条件变分自编码器(VAE)从体密度中生成3D分子。
这些方法实现了分子与口袋几何互补的定向生成,但受限于体素分辨率及旋转不等变性,难以捕捉精细相互作用。
基于图神经网络的生成方法
图模型将原子视为节点、键视为边,可自然表示分子拓扑:
- GraphBP、Pocket2Mol、FLAG 等方法在保留空间信息的同时生成化学合理结构;
- 3DSBDD 利用自回归采样在3D点云上逐原子构建分子;
- GraphBP 通过局部坐标系统实现等变性;
- FLAG 则以片段为单位组装,提高生成速度与可合成性。
这些模型在结合亲和力与分子多样性上显著超越早期方法,成为当前主流框架。
基于扩散模型的生成方法
扩散模型通过逐步去噪从随机噪声生成完整分子,目前成为热点。
典型方法包括:
- ShapeMol:基于形状条件的等变扩散模型;
- MolSnapper:引入药效团约束,结合E(3)-等变神经网络生成高效结合分子;
- DiffSBDD、TargetDiff:在全原子层面扩散坐标与类型,取得较高Vina评分;
- DecompDiff:显式建模分子分区(骨架与支链)以增强生成精度;
- FLOWR、FlexSBDD:使用流匹配(flow matching)替代随机扩散,改进训练稳定性并考虑蛋白柔性。
未来方向
结合特异性评估
当前模型普遍以对接分数代替结合特异性,但其与真实生物结合并不总一致。LiGAN 的突变实验提供了评估特异性的定量框架,表明生成结果依赖于受体结构。未来需建立系统基准测试,以比较不同模型在结合选择性方面的性能。
泛化与偏倚控制
公开蛋白–配体数据库存在采样不均与元数据缺失,易导致模型学习到非物理偏差。研究人员建议:
- 采用跨靶点划分与分子去重;
- 引入多源验证集;
- 利用实验负样本增强判别能力。
结构柔性与动力学
大多数SBDD模型假定蛋白结构静态,而真实结合涉及诱导契合与构象变化。未来趋势包括:
- 联合生成配体与蛋白侧链构象(如FlexSBDD);
- 引入分子动力学或能量约束以提升物理真实性。
多模态融合与可解释性
结合化学语言模型(SMILES)、序列到结构生成网络与图模型的多模态融合将成为新方向。研究人员还呼吁开发可解释机制,以揭示模型如何利用结构信息进行决策,从而促进实验验证与信任。
结论
深度学习正重塑结构基础药物设计的范式。从体素到图,再到扩散与流匹配,模型在分子生成的精度、效率与物理合理性方面持续进步。未来,通过整合蛋白结构、动力学与AI模型的可解释性,研究人员有望实现从结构信息到药物分子的端到端智能设计,加速药物研发的创新周期。
整理 | DrugOne团队
参考资料
Vost, L., Ziv, Y., & Deane, C. M. (2025). Incorporating targeted protein structure in deep learning methods for molecule generation in computational drug design. Chemical Science. The Royal Society of Chemistry.
https://doi.org/10.1039/D5SC05748E
内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-10-20,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号