Nat. Comput. Sci. | 基于自上而下质谱的蛋白数据库蛋白型搜索

Nat. Comput. Sci. | 基于自上而下质谱的蛋白数据库蛋白型搜索

DrugAI
发布于 2026-01-06 12:56:54
发布于 2026-01-06 12:56:54
DRUGONE
研究人员提出了一种新的搜索算法用于蛋白型(proteoform)的鉴定。该方法通过计算蛋白质质量图与谱质量图之间的最大规模误差校正比对,实现了对复杂蛋白型的高效识别。具体而言,研究人员结合了一种筛选算法与搜索算法,使得在不降低精度的前提下,搜索速度比已有方法提升 3.9 至 9 倍。通过构建模拟自上而下质谱数据的管道,该方法在模拟数据集上的准确率达到 95%,在真实注释数据集上的准确率不低于 97.1%。这一成果为大规模蛋白型解析提供了高效、精确的工具。

蛋白型是由基因通过序列变异、可变剪接以及翻译后修饰所产生的不同蛋白质形式, 在生物学过程中扮演着关键角色。自上而下质谱(Top-down MS)技术能够直接分析完整蛋白,揭示其全貌。随着质谱及完整蛋白分离技术的发展,自上而下分析正逐步从单一蛋白拓展到全蛋白组范围,为解析蛋白型功能、发现疾病分子标志物及潜在药物靶点提供了独特优势。
然而,蛋白型的鉴定面临巨大挑战:由于修饰类型和数量的组合爆炸,现有的主流软件工具往往不得不排除大量潜在蛋白型,以降低运行时间。这显著限制了复杂蛋白型的识别能力。
方法
研究人员基于蛋白型质量图(PMG)与谱质量图(SMG)的比对问题,引入了误差校正比对(Error-Correction Alignment, ECA)。该方法不仅考虑实验中峰值的质量误差,还在动态规划中对每个比对峰值进行误差修正,从而避免误差累积。研究人员在此基础上提出了 sTopMG 算法,并结合筛选方法以进一步加速。
此外,为了评估性能,研究人员开发了一条管道,将现有的 MaSS-Simulator(原用于自下而上质谱)改造为自上而下谱模拟器,生成带有不同修饰的模拟谱图,从而在可控条件下检验方法的准确性与效率。
结果
模拟数据
- 研究人员构建了 100 个模拟谱图,并与大肠杆菌数据库进行比对。
- sTopMG 能够在 96 个案例中找到正确的片段,而传统方法 TopMG 仅识别出 50 个,TopMGFast 更低。
- 在准确率上,sTopMG 达到 95%,显著优于现有方法。
真实数据
- 在抗体 Waters 和 HB100 的真实数据集中,使用 FLASHDeconv 解卷积后的谱图,sTopMG 在固定误差容差条件下的准确率达到 97.1%。
- 与主流工具 TopPIC 比较,sTopMG 在多种设置下均表现出更高的正确匹配率。
- 研究人员还发现,解卷积工具的选择对最终结果影响很大,提示解卷积是蛋白型识别中的关键步骤。
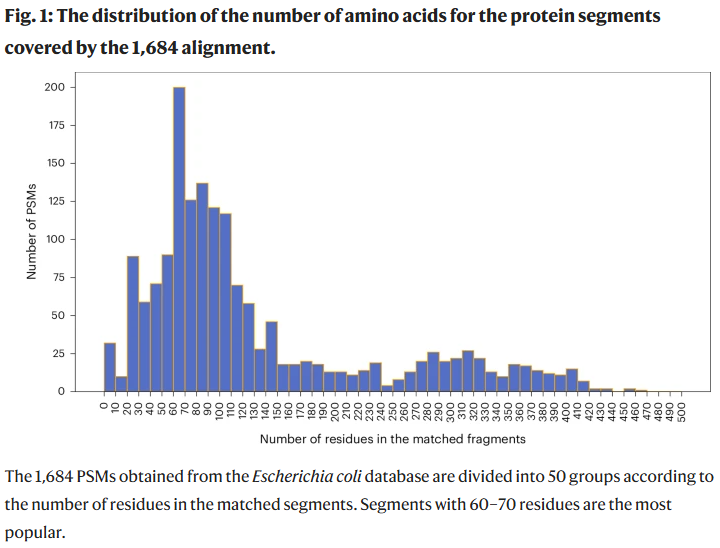
筛选与组合方法
- 新设计的筛选算法可在保持准确率的同时,将运行时间缩短至原来的 1/6。
- 在综合方法(筛选 + sTopMG)中,95% 的模拟谱图得到了正确识别,性能优于 TopMG 与 MS-Align-E 等组合方案。
假发现率控制(FDR)
- 在 Sulfolobus islandicus 的数据集中,研究人员利用目标 + 干扰(decoy)数据库评估假发现率。
- 在 FDR ≤1% 的条件下,sTopMG 始终表现优于 TopPIC。
- 研究人员还分析了不同碎裂类型(ETD vs HCD)对结果的影响,发现 ETD 谱图在鉴定中的表现明显优于 HCD。
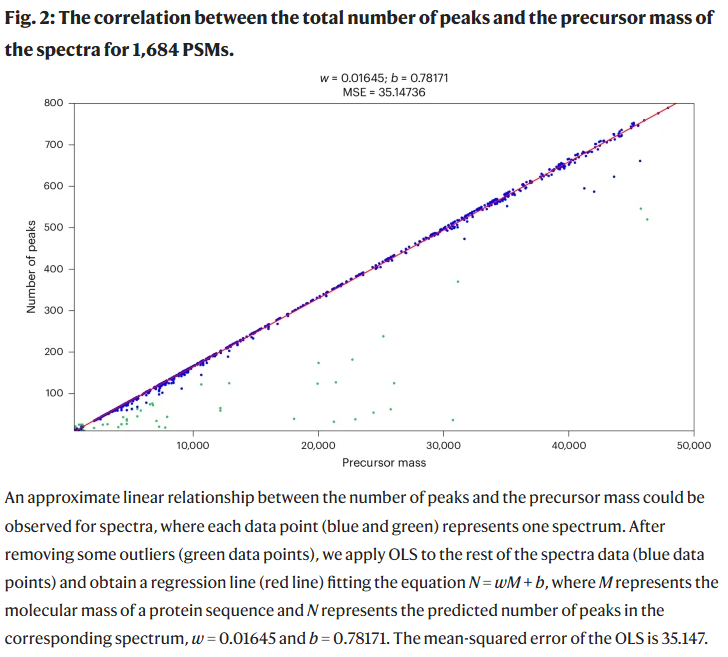
讨论
研究人员展示了一种基于误差校正比对的新型搜索策略,能够在识别复杂蛋白型时兼顾速度与准确性。通过模拟数据与真实数据的双重验证,sTopMG 表现出比现有方法更高的效率和精度。特别是,在抗体和细菌蛋白质组的案例中,该方法证明了其在不同数据集和实验条件下的广泛适用性。
未来的研究需要进一步探索更高效的解卷积方法,并在更多非抗体蛋白数据集上验证其普适性。研究人员认为,该方法为大规模、系统性的蛋白型分析提供了重要工具,并为疾病标志物发现和药物研发奠定了技术基础。
整理 | DrugOne团队
参考资料
Li, K., Shan, B., Xin, L. et al. Proteoform search from protein database with top-down mass spectra. Nat Comput Sci (2025).
https://doi.org/10.1038/s43588-025-00880-z
内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-10-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号