J. Med. Chem. | 基于片段的药物设计:从过去到现在,再展望未来

J. Med. Chem. | 基于片段的药物设计:从过去到现在,再展望未来

DrugIntel
发布于 2025-12-30 20:17:21
发布于 2025-12-30 20:17:21
作者:Weijun Xu and Congbao Kang(原载 《Journal of Medicinal Chemistry》 2025年)
摘要:基于片段的药物设计(FBDD)已成为药物发现领域的一种强大策略,它为传统的高通量筛选(HTS)药物发现方法提供了重要的补充途径。在近半个世纪的时间里,FBDD经历了显著的演变,并成功推动了多个获批药物的问世。结构与计算工具在FBDD中的整合应用,显著提升了其效率,促进了合理的药物设计。随着药物发现领域不断超越传统的可成药靶点界限并探索新的模式,FBDD有望在靶向多种生物分子(包括诸如蛋白质和RNA这类具有挑战性和传统上“不可成药”的靶点)方面发挥关键作用。FBDD的持续进步,特别是通过引入尖端的计算和筛选方法,将为药物化学未来的成功铺平道路。
自1981年首次被提出以来,基于片段的药物设计已证实其作为一种在药物发现项目早期识别苗头分子的有效方法。FBDD涉及筛选低分子量化合物,这些化合物最初与靶蛋白的结合可能较弱,但通过药物化学的努力,有潜力被进一步优化成高效力的药物。随后,在20世纪90年代发展了“通过核磁共振的构效关系(SAR by NMR)”方法,突显了核磁共振(NMR)光谱学在识别小分子片段结合物中的应用。这一策略为FBDD作为药物发现策略奠定了基础(图1)。此后,包括X射线晶体学、表面等离子共振(SPR)和热移分析在内的新方法被开发用于片段筛选,使得基于片段的筛选更易于融入药物发现工作流程。如今,FBDD已成为制药公司、生物技术公司和学术机构广泛使用的主流筛选方法。
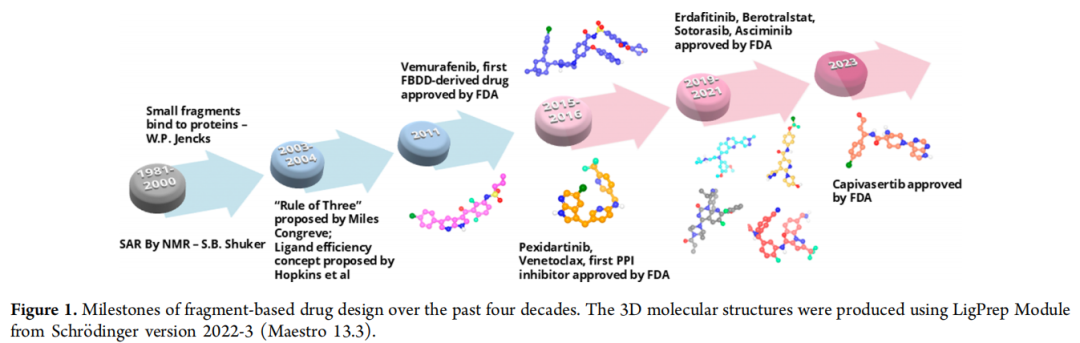
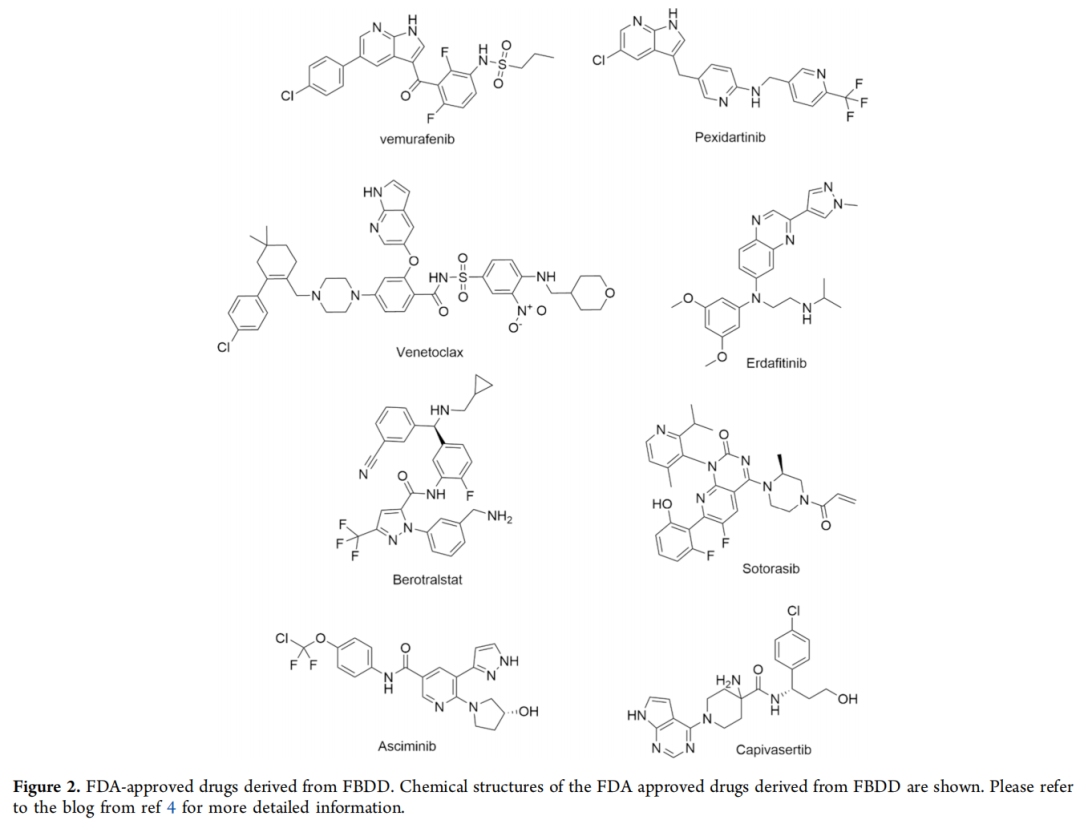
回顾历史,FBDD在药物开发中取得了若干里程碑式的成功(图1),已有八种FDA批准的药物(图2)源于该方法,并有超过50种化合物进入临床阶段。首个源自FBDD的FDA批准药物是Zelboraf(维莫拉非尼,PLX4032),由Plexxikon公司开发的用于治疗黑色素瘤的BRAF抑制剂。该项目于2005年启动,并于2011年获得批准,展示了FBDD在加速药物发现时间线方面的效率。后续受益于FBDD的FDA批准药物包括:Pexidartinib(2015年)-一种CSF-1R抑制剂;Venetoclax(ABT-199, 2016年)-一种破坏蛋白质-蛋白质相互作用的Bcl-2抑制剂;Erdafitinib(2019年)-用于尿路上皮癌的FGFR抑制剂;Berotralstat(BCX7353, 2020年)-用于遗传性血管性水肿的丝氨酸蛋白酶抑制剂;Sotorasib(AMG 510, 2021年)-一种靶向 notorious “不可成药”蛋白KRAS-G12C的抑制剂;Asciminib(ABL001, 2021年)-一种新型的BCR-ABL1变构抑制剂;以及Capivasertib(AZD5363, 2023年)-一种AKT激酶抑制剂。值得注意的是,这些药物中多数属于激酶抑制剂。然而,Venetoclax和Sotorasib的成功凸显了FBDD在针对多种难治靶点药物开发方面的潜力。除了获批药物外,通过FBDD还取得了大量从苗头化合物到先导化合物的成功案例,表明了其在多种靶点家族中的广泛适用性。
在不同的苗头化合物发现筛选方法中,FBDD具有多项优势:它需要筛选的化合物数量相对较少(这些化合物来自多样化的化学空间),使其适用于包括蛋白质和RNA在内的多种靶点,展现出跨靶点的通用性。片段库由低分子量化合物(约150-300 Da)组成,通常遵循“三规则”,为后续优化提供了更大潜力。FBDD采用能灵敏检测弱结合物的筛选方法,这些弱结合物常被高通量筛选所遗漏。有吸引力的是,片段筛选通常可在数天内完成,显著减少了苗头化合物识别所需的时间和成本。此外,片段筛选还能提供关于靶点成药性的初步指示,可作为项目战略规划的风险评估。用户友好的检测设置以及无需参考化合物的特点,使该方法更容易在基于靶点的药物发现项目中实施。在苗头骨架得到验证后,片段生长能够支持系统的构效关系探索,促进新型先导化合物的开发。
超越传统的“五规则”范畴,FBDD也开始在新治疗模式中展现出应用前景,例如蛋白降解靶向嵌合体(PROTAC)。这类分子通常由两个功能部分组成:一个与目标蛋白结合,另一个与E3连接酶结合,通过化学连接子连接。PROTAC的设计策略遵循了片段连接原理,这是FBDD中的一个关键概念。有趣的是,最近研究表明PROTAC的结合部分常表现出类似片段的特征,包括低分子量以及对目标蛋白或E3连接酶的弱内在结合亲和力。这种相似性表明,FBDD可能在优化和扩展新兴药物模式的化学空间方面发挥重要作用,进一步增强其对未来药物发现的影响力。
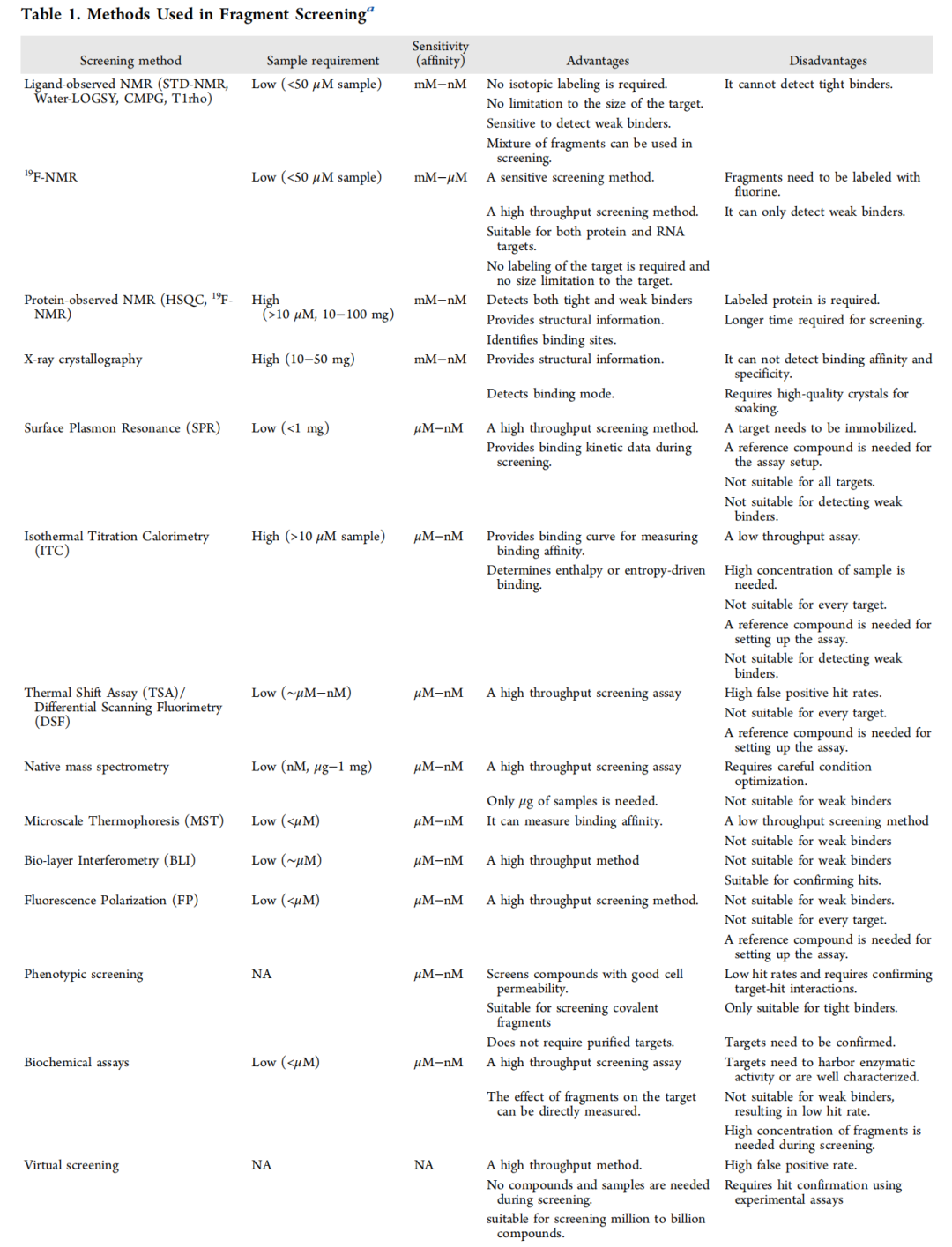
尽管取得了诸多成功,FBDD也面临一些挑战。首先,选择高质量的片段库需要平衡考虑多种因素,包括库的多样性、化学空间覆盖度、理化性质及合成可行性。其次,筛选方法的灵敏度在FBDD中非常重要,因为片段通常与靶点结合较弱。目前有近十几种筛选方法可用,其中NMR是片段筛选和苗头化合物确认的强大工具。配体观测和蛋白质观测的NMR方法能够实现对多样化靶点的高灵敏度片段筛选,并获得令人满意的苗头化合物率。第三,用于片段生长的苗头化合物选择也十分重要。片段筛选通常会在多样化靶点中产生较高的苗头化合物率。配体效率常作为苗头化合物选择过程中的一种“经验法则”,用以反映片段-靶点相互作用的特异性而非泛泛结合。此外,理解苗头化合物的结合模式也是确定优先次序的重要标准。因此,NMR、X射线晶体学和冷冻电镜(Cryo-EM)等结构生物学工具在确定二元或三元复合物结构、在分子水平确认靶点结合方面发挥着关键作用。第四,不断扩大的片段库规模对筛选工作量构成了压力。如今,已存在包含数百万片段的现货库,若不投入大量的时间和资源,很难筛选出高质量的苗头化合物。为克服这一障碍,虚拟筛选等计算方法提供了一种互补解决方案,可在湿实验验证前对超大型片段库进行初筛。另一方面,基于片段筛选获得的苗头化合物可为后续几轮的虚拟苗头化合物优先级排序提供信息,从而实现“计算-实验”迭代循环,提高苗头化合物识别的成功率。最后,片段生长和连接子设计需要药物化学专业知识,以确保新合成的化合物能有效占据蛋白质结合位点。然而,人工驱动的过程本质上受到苗头化合物数量增加以及预测其与靶蛋白相互作用复杂性的限制。因此,可以考虑利用人工智能分析大型数据集,预测蛋白质-配体相互作用,并通过自动化的片段连接和扩展来设计化合物。
未来展望
基于片段的药物设计有潜力成为未来药物发现的关键驱动力。分子生物学和生物信息学的最新进展将靶点空间扩展至RNA和RNA结合蛋白(RBP),为超越传统蛋白质靶点的治疗干预提供了未开发的潜力。例如,FDA批准的RNA剪接调节剂Risdiplam通过改变SMN2前mRNA的剪接,促进外显子7的纳入,从而作为U1小核核糖核蛋白有效剪接其RNA靶点的增强子。然而,RNA和RBP通常呈现高度极性和灵活的结合位点,难以用传统方法靶向。FBDD凭借其识别小而弱结合片段的能力以及对潜在结合位点的更广覆盖,为探索这些复杂靶点提供了一种有效方法。此外,高分辨率的冷冻电镜结构正在阐明复杂的蛋白质/RNA结构,揭示可用于合理药物设计的潜在结合位点。基于人工智能的结构预测工具正在补充实验方法,以帮助理解RNA的三级结构以及RNA与蛋白质之间的复合物结构。先进的分子建模技术提供了关于构象灵活性、配体诱导的结构变化以及可能为片段生长或连接提供机会的潜在隐秘口袋的见解。多样化的片段库,包括共价弹头和特殊基团,正在扩展用于筛选的化学空间。实验与计算筛选方法的结合通过揭示新的可成药位点和提高苗头化合物率,增强了苗头化合物的识别能力。
最终,FBDD与新兴计算技术的协同作用使人们能够更深入地理解作用机制,使得基于片段的方法日益稳健和多功能。随着该领域的不断发展,FBDD有望解决历史上难以处理的靶点,如变构调节位点、蛋白质-蛋白质相互作用、RNA以及RNA结合蛋白。将机器学习和人工智能整合到FBDD工作流程中,有望扩大规模并加速苗头化合物的识别、优先级排序和优化过程,确保更高效的药物发现流程,以满足未竟的医疗需求。
参考文献:Weijun Xu and Congbao Kang, Fragment-Based Drug Design: From Then until Now, and Toward the Future, Journal of Medicinal Chemistry, 2025, 68 (5), 5000-5004
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-12-18,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号