新手小白指南:如何开启单细胞数据分析


今天,我们来读一篇文章,是关于单细胞数据分析入门的。

首先是几个常识概念的介绍。目前单细胞数据分析主要有两大平台:
- • 基于 R 语言的 Seurat
- • 基于 Python 语言的 Scanpy
目前最常用的单细胞测序技术是:10x Genomics。
单细胞数据生成和分析的主要步骤如下图:

数据矩阵生成与质量控制
单细胞分析的一项关键技术进展是条形码技术的发展,该技术可实现大规模并行化,同时将成本降至最低。条形码在逆转录过程中被添加到 RNA 分子上,能够识别单个细胞和独特分子。
单细胞数据分析第一个步骤就是生成表达量矩阵,10x 数据用的软件是 CellRanger。当然还有其他替代工具:kallisto、STAR、STARSolo 等。
表达量矩阵中每个条形码可能代表:
- • 一个单细胞
- • 一个双细胞( doublet )
- • 或一个不含细胞但含有环境 RNA 的 “空” 液滴。
需要指出的一个重要问题是,标准流程会将测序数据与转录组(如经过加工的成熟 mRNA)进行比对。然而,单细胞核 RNA 数据或表观基因组数据(转座酶可及染色质测序技术,ATAC-seq)应与全基因组进行比对,因为细胞核中主要含有前体 mRNA,其中包含内含子区域。原始读数计数通常还会过滤掉在极少数细胞中检测到的基因,从而有效减小数据矩阵的规模。
分析流程的下一步是质量控制(QC),例如:
- • 确定每个条形码的计数数量
- • 每个条形码对应的基因数量
- • 以及每个条形码中来自线粒体基因的计数比例。
基因数量少且线粒体读数比例高通常表明细胞质量较差。但有些细胞(如肾脏近端和远端曲小管细胞)富含线粒体,这是正常情况。异常高的读数和基因计数可能代表双细胞。
控制环境 RNA 污染也至关重要。环境 RNA 是存在于单细胞溶液中并在封装过程中被纳入油滴的 RNA。我们通常使用 SoupX 工具,该工具通过空液滴估算环境 RNA 污染(图 2)。
在典型分析中,我们会考虑多个 QC 参数进行过滤,并迭代使用这些参数。
小编:上游生成表达量矩阵可以使用 CellRanger,STARSolo,以及最新的 kb-python。质控主要考虑的指标有:
- • 细胞总的表达量计数
- • 细胞表达的基因数
- • 细胞中线粒体基因计数占总计数的百分比
- • 双胞
- • 环境 RNA 污染

标准化
单细胞数据需要不同类型和级别的标准化处理(图 1)。例如,总测序读数计数会影响原始计数数量,因此需要根据整体计数深度对基因计数进行缩放。一种常用方法假设每个细胞初始转录本数量相同,简单地将数据标准化为每百万计数。Scran 采用基于池化的大小因子估计和线性回归进行数据标准化,是除 Seurat 使用的简单对数标准化之外最受欢迎的方法之一 ¹⁸。此外,还开发了其他方法,如 SCtransform¹⁹、SCnorm²⁰ 和 BayNorm²¹。标准化后,数据会进行 log (x+1) 转换。通常需要从数据中剔除与细胞周期相关的变异,这一功能已包含在 Seurat 或 Scanpy 的标准分析平台中。该平台还允许剔除其他技术或生物学变异。
小编:大道至简。根据经验,其实最简单的 CPM + log(x+1) 方法通常就是最好的方法。另外留一个问题,我们知道 Bulk 转录组标准化时,要考虑文库大小和基因长度,为什么单细胞标准化通常只考虑文库大小,不考虑基因长度呢?
批次效应校正和数据整合
不同批次的数据要整合到一起,流行的工具有:
- • Seurat 当中的典型相关分析(CCA)或反向主成分分析(RPCA);
- • Scanpy 当中的 Scanorama
而最常用的工具当属:Harmony

可视化和聚类
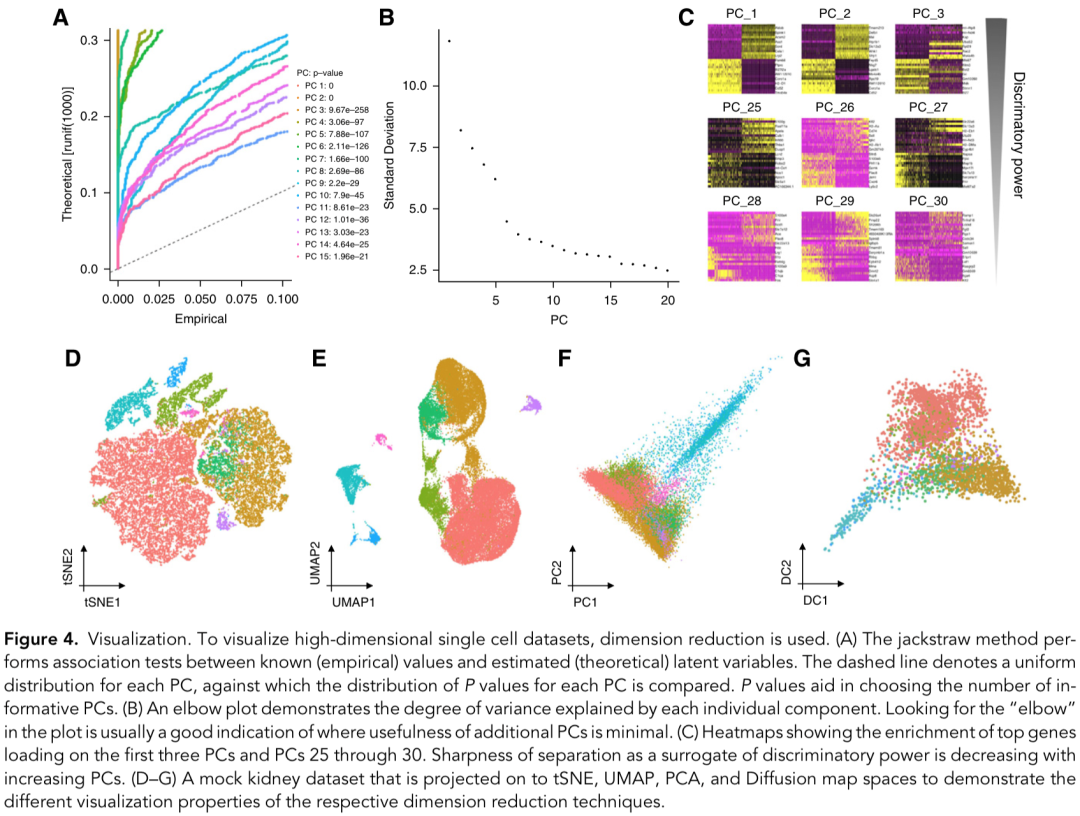
在这一章中,作者提到了使用 PCA 降维,并用 t-SNE 和 UMAP 算法进行可视化。聚类的话,目前Louvain 和 Leiden 最两种最流行的方法。
经典的细胞类型注释使用被视为金标准的外部数据集。用于肾脏细胞类型注释的外部数据集越来越多,包括 Susztak 实验室 ³²⁻³⁵、Humphreys 实验室 ³⁶⁻³⁷、Tabula Muris³⁸、人类细胞图谱(Human Cell Atlas)³⁹、肾脏上皮细胞本体论网页 ⁴⁰⁻⁴² 和免疫基因组计划联盟(ImmGen Consortium)⁴³。
最近,自动化细胞注释工具得到了发展,例如 Garnett⁴⁴(预印本)、SingleR⁴⁵、CHETAH⁴⁶ 和 MOANA⁴⁷,这些工具提供了更全面、更具概率性的细胞身份注释方法。同一细胞类型的标记基因在不同数据集之间可能存在差异。

细胞水平分析:细胞比例变化、分解和轨迹分析
细胞比例的变化(数据集中每种细胞类型的比例)与疾病状态密切相关,这是单细胞分析最简单的输出结果之一。
这些数据可以提供不同条件之间的相对估计,但由于单细胞文库制备过程中细胞捕获存在偏差,从单细胞数据中推断出的细胞比例可能不准确。
仅通过聚类这种离散分类系统无法充分描述细胞多样性。轨迹分析可捕捉细胞在过渡过程中的显著特征,例如器官在多个时间点的发育过程、疾病状态之间的转变、细胞历史或拓扑信息。
因此,捕捉细胞身份之间的转变、分支分化过程或生物学功能的渐进式、非同步变化,需要基因表达的动态模型。Monocle 是一种机器学习方法,用于重建每个细胞从一种状态过渡到另一种状态时必须执行的基因表达变化序列
最近开发的 RNA 速率分析(如 velocyto 软件包中实现的方法)是一种分析细胞历史的新方法 ⁵⁷。RNA 速率是基因表达状态的时间导数,可以通过在常见的 scRNA-seq 方案中区分未剪接和已剪接的 mRNA 来直接估计 ⁵⁷⁻⁵⁸。
基因水平分析:差异表达、基因调控网络、驱动通路和细胞间相互作用
差异表达(DE)分析是在未校正的数据上进行的,分析过程中会纳入技术和生物学协变量。Seurat 使用不同的模型进行 DE 分析(图 6)。
基因水平分析还可以与基因集富集分析方法相结合,例如基因集富集分析(GSEA)或加权基因共表达网络分析(WGCNA)⁶⁴。
为了解释 DE 结果,我们通常根据基因参与的共同生物学过程对其进行分组。生物学过程标签存储在多个数据库中,例如 MSigDB⁶⁵、基因本体论(GO)⁶⁶⁻⁶⁷、京都基因与基因组百科全书(KEGG)⁶⁸ 和 Reactome⁶⁹ 数据库。尽管需要注意的是,某些通路成员的基因表达富集并不一定与通路活性相关,但可以使用多种工具对基因列表上的注释富集进行检验,这些工具在其他文献中已得到综述和比较 ⁷⁰⁻⁷¹。
单细胞分析领域的一项最新进展是使用配对基因标签进行配体 - 受体分析 ⁷²。在此分析中,细胞簇之间的相互作用是通过受体及其同源配体的表达来推断的。配体 - 受体对标签可从最新数据库(如 CellPhoneDB⁷³ 或 Connectome⁷⁴(预印本))中获取,并通过统计模型用于解释不同簇间高表达的基因 ⁷⁵⁻⁷⁷。

单细胞分辨率下的基因调控
单细胞核转座酶可及染色质测序(snATAC-seq)通过分析染色质可及性,允许在单细胞水平上分析表观基因组图谱(图 7)。已开发了多种用于 snATAC-seq 分析的工具,最著名的包括 Ren 实验室开发的 SnapATAC⁷⁸(预印本)、Satija 实验室开发的 Signac⁷⁹(预印本)和 Greenleaf 实验室开发的 ArchR⁸⁰(预印本)。我们倾向于使用 SnapATAC,这是一种非线性维度约简方法。

网络工具和数据集
在过去几年中,已经生成了大量人类和小鼠肾脏数据集。原始数据集通常可从基因表达综合数据库(GEO)下载。大型综合人类肾脏参考注释将作为人类细胞图谱项目 ³⁹ 和人类生物分子图谱计划(Human BioMolecular Atlas Program)⁸⁶ 的一部分提供。肾脏精准医学项目(Kidney Precision Medicine Project)⁸⁷(预印本)旨在生成多种人类肾脏疾病状态的数据集。肾脏重建联盟(Rebuilding a Kidney consortium)⁸⁸ 将分析发育中的人类肾脏样本和体外分化的肾脏类器官。
下面列举一些资源:
- • Humphreys 实验室的 KIT 网站允许快速可视化其大量数据(http://humphreyslab.com/SingleCell/)
- • McMahon 和 Kim 实验室使用 VisCello 可视化发育中和成年小鼠的数据,并比较雄性和雌性动物之间的差异:http://susztaklab.com/VisCello/
- • Satija 实验室开发的独立分析应用程序(http://azimuth.satijalab.org/app/azimuth)
空间和多组学数据集
关于空间和多组学数据集整合这一新兴领域,我们建议读者参考最新的优秀综述 ⁹¹⁻⁹⁴ 以及补充材料 1-3。
结论
目前,肾脏疾病根据其病程(如急性或慢性)或组织学描述进行分类,而组织学描述是基于几个世纪前提出的颜色和形状同源性。这些描述无法捕捉疾病驱动分子通路的潜在分子机制,因此不适合用于靶点识别和药物开发 ⁹⁵⁻⁹⁶。单细胞技术可以解析疾病状态的变化,实现新的分子疾病分类,并有助于潜在靶点的识别。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-11-25,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号