汤富酬团队新作:利用AI对scRNA-seq数据注释和多数据集整合

汤富酬团队新作:利用AI对scRNA-seq数据注释和多数据集整合

生信菜鸟团
发布于 2025-07-24 15:19:23
发布于 2025-07-24 15:19:23
2025 年 6 月北京大学汤富酬团队发表在 Genome Biology 上的《scExtract: leveraging large language models for fully automated single-cell RNA-seq data annotation and prior-informed multi-dataset integration》利用大语言模型实现了完全自动化的 scRNA-seq 注释以及基于先验信息的多数据集整合。
内容精简版:
自 scRNA-seq 技术出现以来,公开可用的单细胞测序数据量持续迅猛增长。截至目前为止,cellxgene 作为最大的文献整理单细胞数据库,已包含 1844 个数据集,主要是人类和小鼠的单细胞 RNA 测序数据。而一年新增的数据集就有数千个,这些分散的数据集涵盖了从基本细胞过程到多种人类疾病(包括癌症、神经退行性疾病和免疫疾病)的研究。然而,有效利用这些公共数据却面临巨大挑战:
1)注释缺失:原始数据常缺乏细胞类型标签,手动查阅文献提取预处理参数、聚类信息和标记基因耗时费力;
2)自动化瓶颈:现有自动注释工具(如 SingleR)依赖参考数据集,难以发现新细胞类型,对罕见病亚型尤其无力;
3)整合失真:传统批次校正方法(如 Scanorama)在合并多数据集时,容易模糊生物学差异或丢失稀有细胞群。这些问题严重阻碍了大规模细胞图谱的构建和跨研究的生物学发现。
scExtract 通过大语言模型(LLM)驱动的全自动化框架系统解决这些挑战:
- 通过 LLM 自动阅读研究论文 PDF,提取关键实验参数和生物学背景知识,实现与原文一致的预处理和聚类。
- 结合提取的文献信息和数据本身,进行两轮自动化细胞类型注释。首轮基于标记基因和背景知识生成标签,次轮通过查询关键基因表达进行自我验证与优化,显著提升罕见细胞识别精度。
- 创新性地开发了
Scanorama-prior和Cellhint-prior算法。它们将自动化注释作为“先验知识”融入整合过程:Scanorama-prior在计算细胞距离时加权细胞类型相似性,更好地保留生物学结构;Cellhint-prior则根据注释不确定性动态调整先验权重,提升对命名误差的鲁棒性。两者协同,实现了高保真的跨数据集整合。
scEXtract 处理一个数据集仅需 20 分钟,成本不足 1 美元(基于 Web API),彻底解放人力。随着 LLM 发展,精度和效率将持续提升。本文利用 scExtract 成功整合了 14 个异质皮肤数据集(44 万细胞),构建首个覆盖多疾病状态的人类皮肤免疫图谱,并发现银屑病特异的 CXCL14+ 增殖性角质细胞亚群。scExtract 为单细胞组学提供了强大的基础设施,支持快速构建定制化参考图谱,极大加速跨数据集 Meta 分析和新颖生物学发现的进程。
scExtract 基本原理
scExtract 输入:原始 count 水平的数据集(.h5ad 格式)+ 研究论文(PDF 格式)
数据集处理流程:(下图左侧)
- 细胞过滤:去除低质量的细胞。默认参数为细胞中至少包含 300 个基因,仅当论文中明确提及时才应用额外阈值。
- 聚类前预处理:大多数参数使用默认设置。关键可调参数包括:高变基因数量、kNN 图邻域大小(
n_neighbors)、是否使用harmonypy进行样本级批次校正等。 - 聚类:方法选择
Leiden或Louvain,从论文中直接提取明确声明的细胞簇数量,未明确声明时根据文本描述推断,基于推断的聚类数自动优化分辨率参数。 - 首轮注释:通过 Wilcoxon 秩和检验筛选每个簇中 top10 高变基因,LLM 结合背景知识对细胞簇进行注释,输出细胞类型、置信度、组织来源等。由于 LLM 输出存在固有差异性,这些注释结果需要谨慎解读。
- 次轮注释(可选):scExtract 允许 LLM 自主选择基因,将聚类层面的表达数据通过工具模型转化为自然语言,再输入 scExtract 进行判断和重新注释,以解决低置信注释。
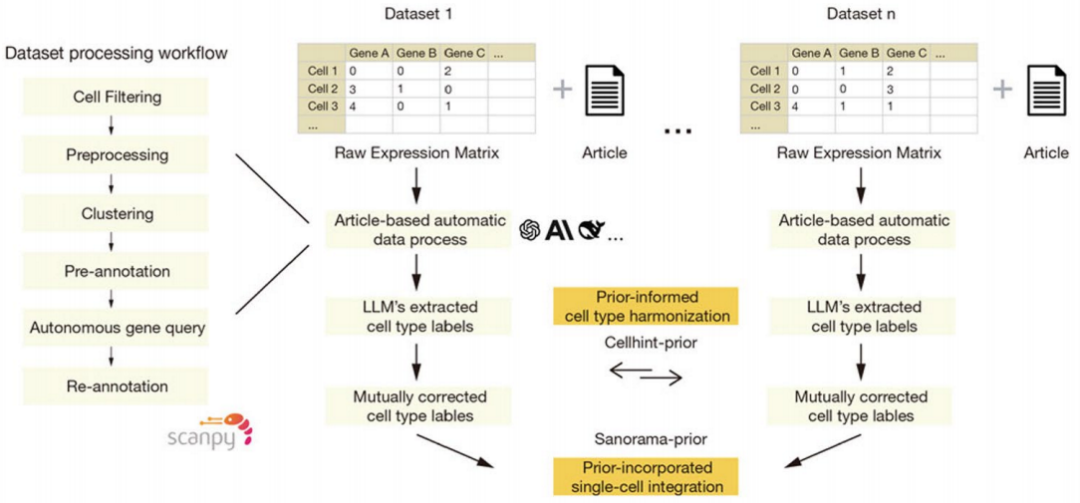
Cellhint-prior 和 Scanorama-prior 算法的理解就没有那么轻松,在此之前先了解 Cellhint 和 Scanorama 算法帮助更好地理解:
Scanorama:基于互近邻(MNN)的批次校正
不同单细胞数据集因技术差异(如测序平台)产生“批次效应”,导致相同细胞类型在降维图中分离。在基因表达高维空间中,若细胞A(数据集1)的最近邻居是细胞B(数据集2),且细胞B的最近邻居也是细胞A,则A-B构成互近邻对(MNN pair),代表同一生物学状态。每个MNN对生成一个位移向量(细胞A到B的方向)。所有MNN向量的加权平均决定整个数据集的整合方向。
Scanorama 依赖表达相似性,若批次效应强或稀有细胞少,MNN配对可能错误,导致过度校正(抹杀生物学差异)或校正不足(批次残留)。
Scanorama-prior 的关键改进:
1. 改进 MNN 的搜索:
引入细胞类型相似性矩阵 MIJ,该矩阵通过计算不同细胞类型标签的文本嵌入向量之间的余弦相似度得到。原始的 Scanorama 计算细胞距离 dij 仅基于基因表达,Scanorama-prior 将原始的基因表达距离 dij 根据细胞 i 的注释标签 I 与细胞 j 的注释标签 J 的相似度 MIJ 进行修正:
如果细胞 i 和 j 的类型高度相似(MIJ 接近 1),则修正后的距离 d‘ij 几乎等于原始距离 dij ,不影响它们成为 MNN 的可能性。如果细胞 i 和 j 的类型不相似 (MIJ 很小),则修正后的距离 d‘ij 会变得很大,这大大降低了它们被选为 MNN 对的可能性。
2. 改进细胞的位移校正:
在找到 MNN 对并计算了基础偏移向量后,Scanorama 使用这些向量来移动批次内的细胞。 Scanorama-prior 增加了一个基于细胞类型中心位置的校正项:
Biasij :针对细胞 i 和 j 所组成的MNN 对 Scanorama 计算出的原始偏移向量。
vi :细胞 i 的坐标向量。
vI :细胞 i 所属细胞类型 I 的所有细胞的平均坐标(中心)。
vj :目标批次中与 i 配对的 MNN 细胞 j 的坐标。
vJ :细胞 j 所属细胞类型 J 的中心。
(vi - vI):表示细胞 i 相对于其自身类型中心的位置偏移(内部结构)。
(vj - vJ):表示细胞 j 相对于其自身类型中心的位置偏移。
(vi - vI - vj + vJ ):这个向量表达了细胞 i 和 j 相对于各自类型中心位置的相对差异。
(vi - vI - vj + vJ )这个额外偏移量被细胞类型相似度 MIJ 加权。当 i 和 j 的类型高度相似(MIJ 接近 1)时,这个额外偏移量被强烈应用。它不仅把细胞 i 移向 j,还努力让 i在整合后的新空间中,相对于其新类型中心的位置,保持与 j 相对于其类型中心的位置 相同的相对关系。当 i 和 j 的类型不相似(MIJ 接近 0)时,这个额外偏移量几乎不起作用,主要依赖原始的 Biasij 进行校正。
Cellhint:基于层级聚类的细胞类型对齐
不同数据集对相同细胞类型命名不一致(如"CD4+ T cell" vs "Helper T"),阻碍了跨数据集的比较,因此开发了 Cellhint 用于跨数据集细胞类型的对齐与协调。Cellhint 输入多个数据集及其各自的细胞聚类结果和注释,计算所有数据集间所有细胞类型(或聚类)两两之间的相似性,构建一个包含所有细胞类型节点的全局相似性图,然后应用 PCT(一种多输出回归算法)对不同数据集的细胞类型进行对齐,最终输出一个协调后的、跨数据集一致的细胞类型注释方案。
Cellhint 需要预先的细胞聚类和注释,对单细胞噪声敏感,而且对齐结果依赖于输入的聚类质量和相似性计算。
Cellhint-prior 的关键改进:
将细胞类型注释的语义信息(文本嵌入向量)和每个细胞注释的置信度引入到 Cellhint 计算细胞类型相似性的核心步骤中。
βααβ
SiJ :细胞 i 与细胞类型 J 通过 Cellhint 计算得到的相似度。
αi :细胞 i 的注释置信度。
β :一个超参数(论文中默认设为 0.1),控制先验信息整体影响的强度。
当细胞 i 置信度低(αi 小)时,修正后的相似度 S'iJ 主要依赖于原始的表达谱相似度 SiJ ,先验知识 MIJ 的贡献很小;当细胞 i 置信度高(αi 大)时,可以更多利用 I 和 J 的语义关系 MIJ 来指导细胞类型的对齐。
scExtract 性能评估
1. 评估 scExtract 聚类和注释准确性
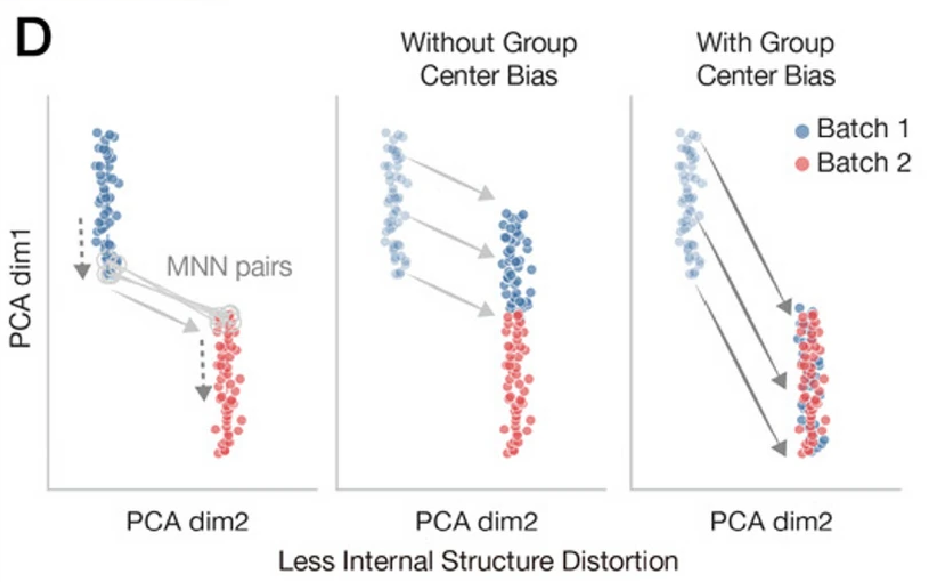
图2A 为评估流程示意图。 通过 18 个来自 cellxgene 数据库的中等规模数据集(21 个数据集里提出了 3 个没有独特细胞类型的数据集),对比了 scExtract(使用三种 LLM API:DeepSeek-v2.5、GPT-4o-mini、Claude-3.5-sonnet)与其他主流自动注释方法(SingleR、scType、CellTypist)的性能。将自动注释结果与人工注释的“金标准”进行比对。
图2B 为原始文献报道的真实细胞类型数量和自动注释方法预测的细胞类型数量的 pearson 相关性图。图2C 为调整兰德指数(Adjusted Rand Index, ARI)条形图,ARI 用于衡量自动注释与标准注释的聚类结构一致性,0~1之间,越高越好。Group based 表示先对细胞进行无监督聚类,将细胞划分成不同的 cluster,然后对整个细胞群体进行统一注释;Single-cell based 表示直接对每个单独的细胞进行分类注释,无需先进行聚类分组。图2D 使用 text-to-embedding 方法,将细胞类型名称的文本描述,通过 LLM 转换为向量,计算自动注释结果与人工金标准之间的余弦相似度。图2E 表示各方法中不同丰度细胞类型的注释准确性。图2F 表示使用背景知识能够显著提升注释准确性。图2G 表示 scExtract 在不同置信度水平下的注释准确性。图2H-I 分别表示四种方法的 text-to-embedding 相似度和 cell ontology 相似度。
图2J 为肾脏数据集的可视化,表格左边为人工注释的金标准,右边为 scExtract(Claude 3.5)自动注释结果。图2K-M 分别是使用 scExtract 和 SingleR 进行自动注释的可视化结果和与金标准的注释相似性。图2N 为 scExtract 第二次重复运行的注释结果,首次运行标记为"Unknown"的细胞亚群被正确注释。
总结一下这部分的内容就是:scExtract 在跨组织数据集上可以实现高精度注释,优于现有方法,且对稀有细胞类型更具鲁棒性。使用背景知识(文献)以及二轮注释可以有效提升注释结果的准确性,且 scExtract 的置信度评分能够反映实际准确性。
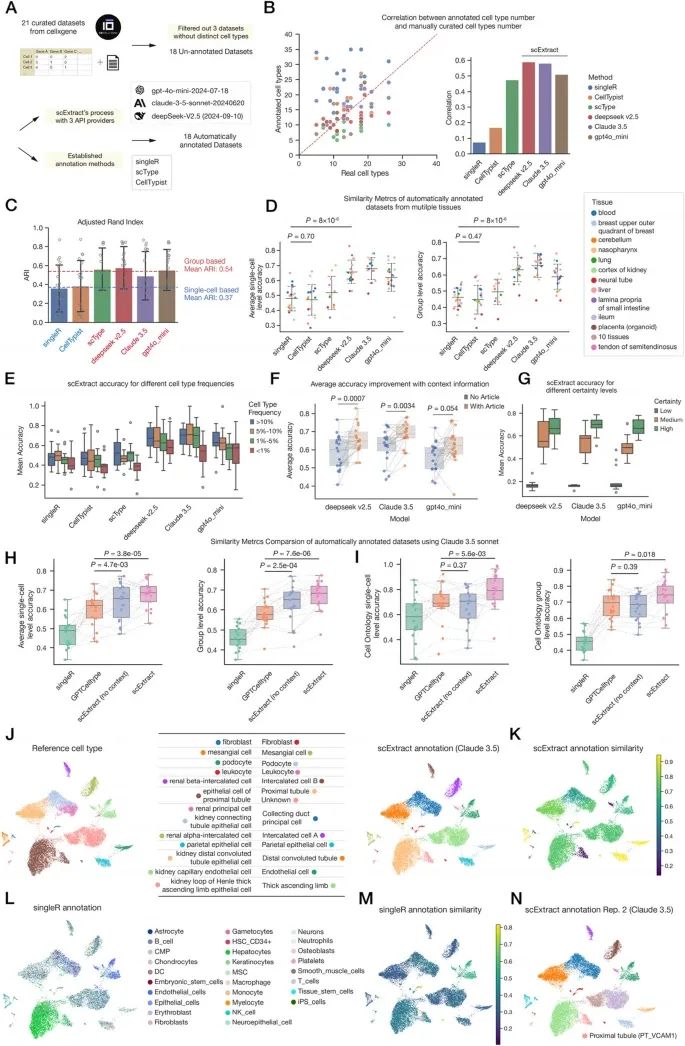
2. Scanorama‑prior 和 Cellhint‑prior 增强了跨数据集整合能力
图3A 为不同测序平台的人类胰腺数据集 scanorama vs scanorama-prior 整合结果可视化比较,上排是 scanorama 整合结果,下排是 scanorama-prior 整合结果,从左到右不同颜色分别代表不同测序批次、不同细胞类型、Leiden 进行聚类的结果。图3B 是具体的量化指标对比表。主要包含两类:生物学差异保留能力和批次效应去除能力,各指标值越接近 1 结果越好。
图3C-D 分别为 cellhint 和 cellhint-prior 的细胞类型对齐树图。每一列代表一个独立的 batch,每个点代表一个细胞亚群,水平线连接不同数据集中被判定为相同细胞类型的群,线的粗细代表置信度的高低。原始 cellhint 仅能成功对齐 inDrop 内部批次,引入先验知识后所有平台的同类细胞被正确对齐。图3E 为 cellhint vs cellhint-prior 可视化结果的比较。原始 cellhint 批次间仍有明显分离,cellhint-prior 不同批次同类型细胞完全混合。
3. 评估 scExtract 在大规模数据集上两步整合策略的性能
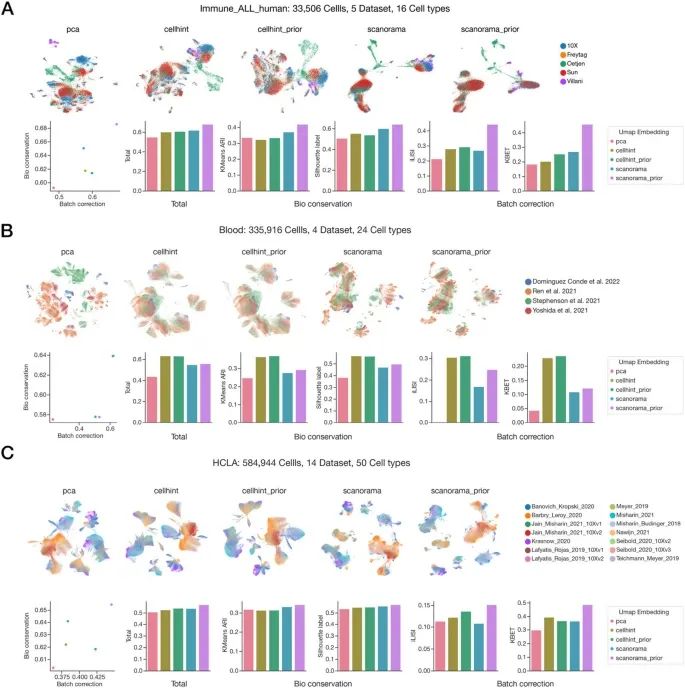
图4 对免疫细胞、血液细胞、人类肺细胞三类细胞进行大规模数据集的整合,上排为 UMAP 可视化对比不同整合方法的结果,下排为量化指标,从左到右依次为:散点图展示批次效应去除和生物学差异保留之间的平衡;柱状图分别呈现综合性能指标、KMeans ARI、Silhouette Label、iLISI 和 kBET,其中 KMeans ARI 反映聚类与真实类型一致性;Silhouette Label 代表同类细胞聚集紧密度;iLISI 表示批次混合度;kBET 反映批次随机分布程度。
总的来说,方法性能依赖数据规模特征,细胞量适中(~ 5 万)以及超大规模 + 多数据集(>10)时推荐使用 scanorama-prior 方法(图4A 和 图4C),效果最好;高细胞量 + 低数据集数目时更建议用 cellhint-prior(图4B)。scExtract 提供了两步整合策略(cellhint-prior → scanorama-prior),可根据数据特征跳过第二步。
4. Case Study:利用 scExtract 自动化构建人类皮肤免疫失调图谱
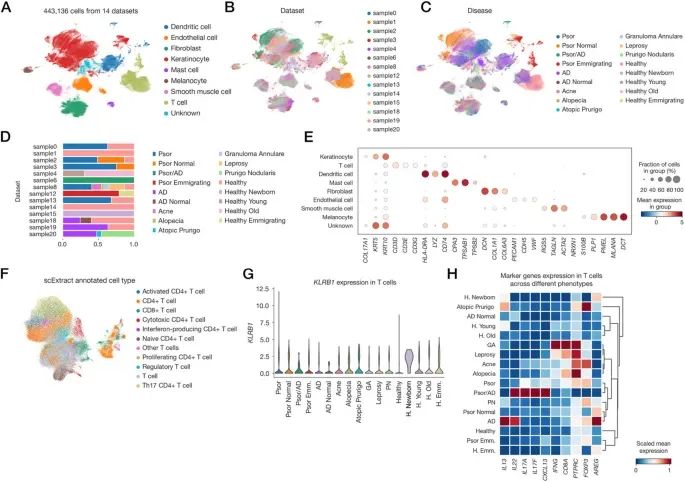
通过完全自动化流程(无人工干预)整合 14 个皮肤 scRNA-seq 数据集(涵盖健康、银屑病、特应性皮炎等状态)。
图5A-C 为整合数据集 UMAP 可视化概览,不同颜色分别代表不同的细胞类型、批次、疾病状态和年龄标签。图5D 展示了不同原始数据集中标签分布比例。图5E 用点状图说明不同细胞类型中标记基因的表达水平,自动注释结果与已知生物学知识一致。
图5F-H 为 首张跨疾病皮肤 T 细胞图谱分析。图5G 中,KLRB1 作为先天淋巴细胞(ILC)的经典标记基因,在新生儿皮肤中高表达,印证胎儿期先天免疫主导,银屑病 ILC 比例低于健康皮肤。图5H 量化了 T 细胞亚群中关键炎症因子的表达水平。
5. scExtract 揭示了不同皮肤疾病中角质形成细胞状态的异质性
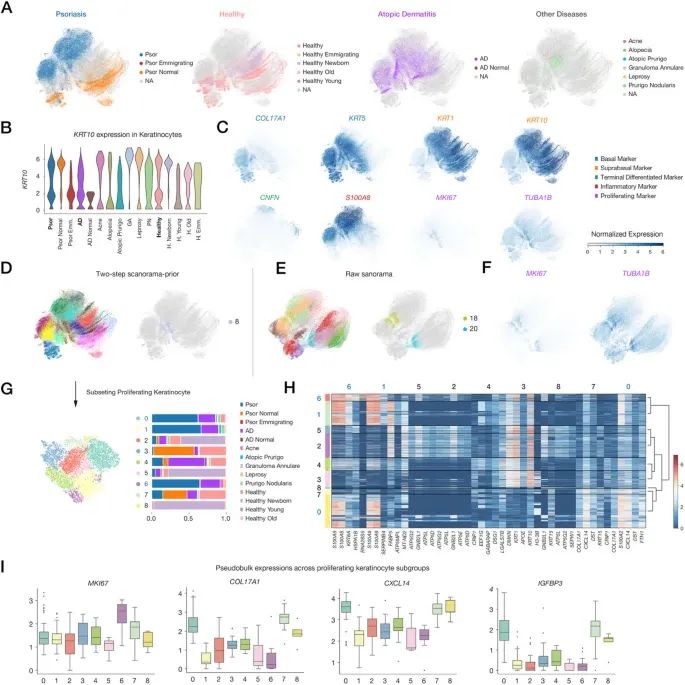
图6A 通过 UMAP 降维展示不同疾病类别(银屑病、健康对照、特应性皮炎、其他皮肤病)中角质形成细胞的分化轨迹。图6B 为角质形成细胞亚群中 KRT10(终末分化标志物)的表达水平。特应性皮炎(AD)细胞中表达 KRT10 的比例较高,但表达强度低于其他疾病。图6C 可视化了特定标记基因的表达,如 MKI67 为增殖标志,S100A8 为炎症标志。
图6D-E 展示了整合方法对增殖性角质形成细胞聚类的影响,D 和 E 分别为使用两步 scanorama-prior 和原始 scanorama 进行聚类的结果,图右侧是识别出的高增殖角质形成细胞亚群,原始方法因批次效应校正不足,将同一增殖亚群错误拆分为两个独立簇。图6F 为在原始 scanorama 整合的 UMAP 上可视化增殖基因的表达,表达模式被干扰,无法清晰识别单一增殖亚群。图6G 为高增殖亚群在不同疾病中的比例。图6H 为增殖性角质形成细胞亚群的差异基因热图。图6I 比较了 MKI67、COL17A1、IGFBP3、CXCL14 在增殖亚群中的表达水平,cluster0(银屑病)中 CXCL14(通常具有抗炎作用)高表达。
图6 和核心结论就是,scExtract 的先验整合(scanorama-prior)能准确校正批次效应,揭示真实生物学差异,发现了银屑病特异的增殖亚群(cluster0),银屑病中 CXCL14 的表达模式可能是一种代偿性抗炎反应。
总结
scExtract 的核心价值在于通过 LLM 桥接生物学知识与计算流程,解决了单细胞数据分析中耗时的手动预处理问题,并为大规模数据整合提供了新范式。然而,其性能仍受限于 LLM 的可靠性、注释标准化缺失及复杂生物学场景的解析深度。未来需结合领域专用 LLM 微调,并扩展至多组学分析以提升普适性。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-07-18,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号