空间转录组学习笔记 - 第3周(初来乍到的Echo)


在空间转录组学领域,技术的发展日新月异,多种技术百花齐放,这些技术之间有何差异,不同平台之间有怎样的区别,也非常值得关注。2024年7月发表在《Nature Methods》上的 Systematic comparison of sequencing-based spatial transcriptomic methods 系统地比较了11种基于测序的空间转录组技术,评估了每种技术在空间分辨率、捕获效率和分子扩散方面的性能,可惜的是10x Visium HD的测评并未被纳入此项研究。
一、技术分类与样本选择
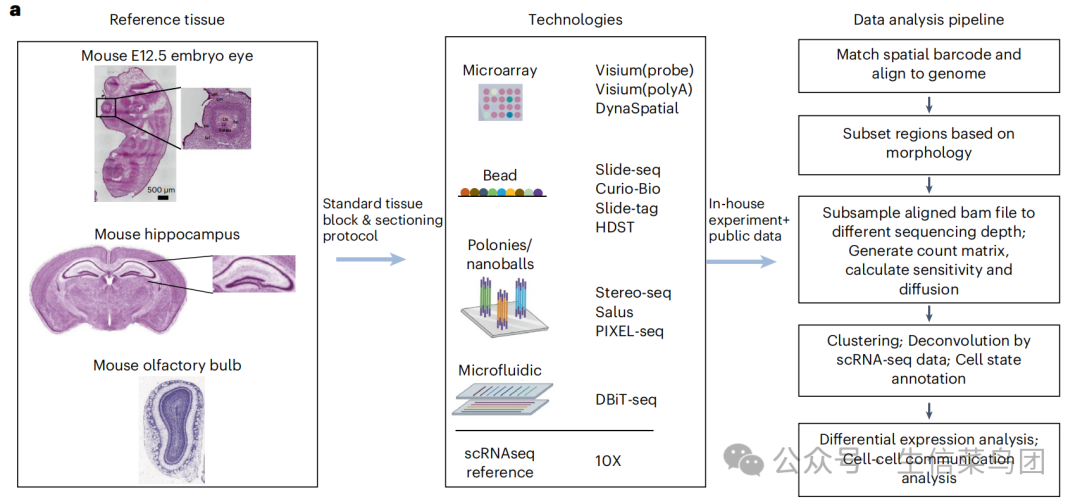
作者系统地将11种空转技术分为四个大类,分别为:
- 基于微阵列芯片的技术(Microarray):Visium(probe)、Visium(polyA)、DynaSpatial
- 基于微珠的技术(Bead):Slide-seqV2、Curio-Bio、Slide-tag、HDST、BMKMANU S1000
- 基于polony或纳米球的技术(Polony/Nanoball):Stereo-seq、Salus、PIXEL-seq
- 基于微流控的技术(Microfluidics):DBiT-seq
作者选择成年小鼠大脑、E12.5 小鼠胚胎和成年小鼠嗅球作为参考组织,因为它们具有相对明确的形态特征。成年小鼠海马体具有稳定的厚度,并包含诸如海马角和齿状回等区域,每个区域都有独特的表达谱。E12.5 小鼠胚胎的眼睛具有已知的结构,即晶状体被神经视网膜细胞环绕,而小鼠嗅球则具有清晰的层分离和各种神经元类型。这些组织因其已知的形态模式和异质性表达谱,成为空转技术基准研究的理想参考样本。
接下来先简单了解一下这四类技术的基本原理:
1. 基于微阵列芯片(Microarray)的技术
基于microarray的技术主要有10x Genomics公司的Visium技术,包括基于探针的方法和基于polyA尾的方法(还有未被纳入研究讨论的Visium HD),以及DynaSpatial。
Visium技术的原理在第一周的笔记中就被介绍了,详细可参考空间转录组学习笔记 - 第1周(初来乍到的Echo)。简单来说就是首先将组织切片固定染色后置于空间芯片上,该芯片上的捕获区域含数百万个独特寡核苷酸探针,组织切片中的mRNA与探针结合实现捕获,随后原位逆转录,使cDNA带上探针中的空间位置信息和UMI信息,将带有信息的cDNA洗脱后进行文库构建,再通过高通量测序平台测序,最后借助生物信息学分析,依据空间条形码确定转录本在组织切片中的位置,结合UMI完成基因表达定量分析,从而获取组织中基因表达的空间分布信息,实现将基因表达数据与组织形态学信息相结合,能在组织原位对基因表达进行高通量检测,并定位基因表达的空间位置。
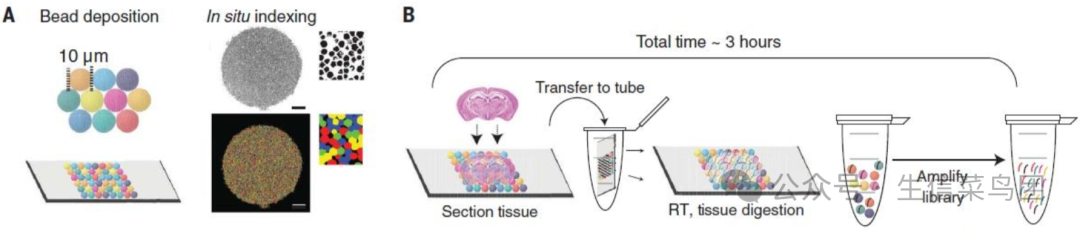
2. 基于微珠(Bead)的技术
基于bead的技术主要有HDST、BMKMANU S1000、Slide-seq V2、Curio Seeker(Slide-seq在Curio Bioscience公司的商业化版本)、Slide-tag。这类技术的基本原理是,通过微米级微珠阵列在组织切片中捕获RNA分子,并利用微珠携带的“空间条形码”将基因表达信息映射回原始位置。
首先,预先制备的每个微珠表面固定有独特的寡核苷酸序列,包含两部分关键信息——空间条形码(对应微珠在阵列中的坐标位置)和唯一分子标识符(UMI)。这些微珠通过微流控、光刻或自组装技术排列成高密度阵列(每平方厘米数万至百万个微珠),形成一张捕获RNA的“坐标网格”。随后,新鲜或冷冻的组织被切成薄片(通常5-20μm),贴合到微珠阵列表面。通过化学透化处理破坏细胞膜,释放细胞内的RNA分子。这些RNA扩散到周围微珠附近,通过微珠上的探针与RNA结合,从而被捕获。每个微珠仅捕获邻近细胞释放的RNA,由此保留空间位置信息。接下来,捕获的RNA分子被逆转录为cDNA,并在过程中将微珠上的空间条形码和UMI整合到cDNA序列中。经过扩增后构建文库,通过高通量测序(如Illumina平台)同时读取RNA序列、空间条形码和UMI。测序完成后,利用预先记录的微珠坐标信息,将每个RNA分子映射回组织中的原始位置,并通过UMI校正PCR扩增偏差,最终生成高分辨率的空间基因表达图谱。
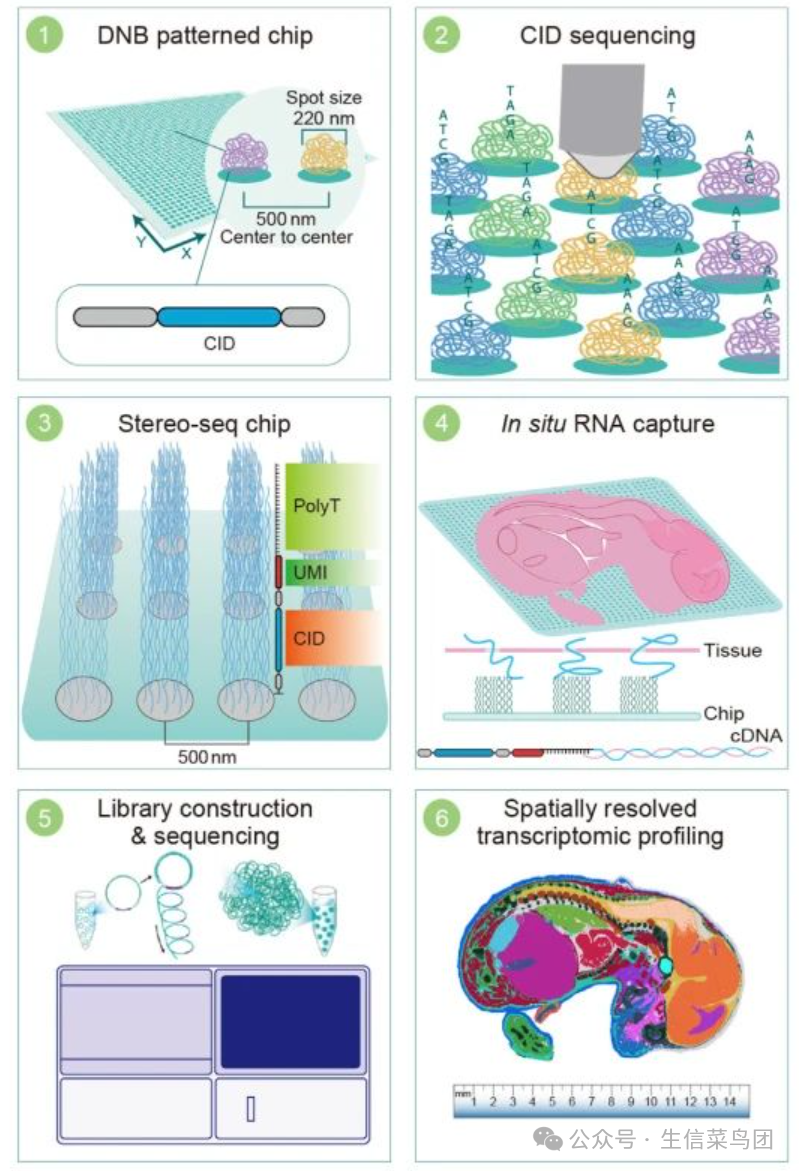
3.基于纳米球(Polony/Nanoball)的技术
基于纳米球的技术主要有Stereo-seq、Salus、PIXEL-seq,其核心原理是通过预固定在载玻片上的DNA纳米球阵列(每个纳米球代表一个“像素点”),结合组合条形码策略,在单细胞或亚细胞分辨率下捕获并定位组织中的RNA分子。
该技术首先将含有随机条形码序列的DNA纳米球(DNB)沉积到经光刻蚀刻的经修饰的芯片上。与基于微珠的方法相比,使用滚环扩增放大产生的标记为DNB的随机条形码取得更大的空间条形码池,同时保持序列保真度。然后对阵列进行显微照相,用引物孵育并测序,以获得包含每个DNB的坐标编码(CID)的数据矩阵。通过与CID杂交,在每个点上连接分子编码(MID)和含有寡核苷酸的polyT序列。下一步包括组织polyA尾RNA的捕获,通过将新鲜氮气冷冻组织切片加载到芯片表面,然后进行固定、渗透,最后进行逆转录和扩增。收集扩增后的cDNA,作为制备文库的模板,与CID一起进行测序。对测序数据进行计算分析,可以实现空间分辨的转录组学研究。
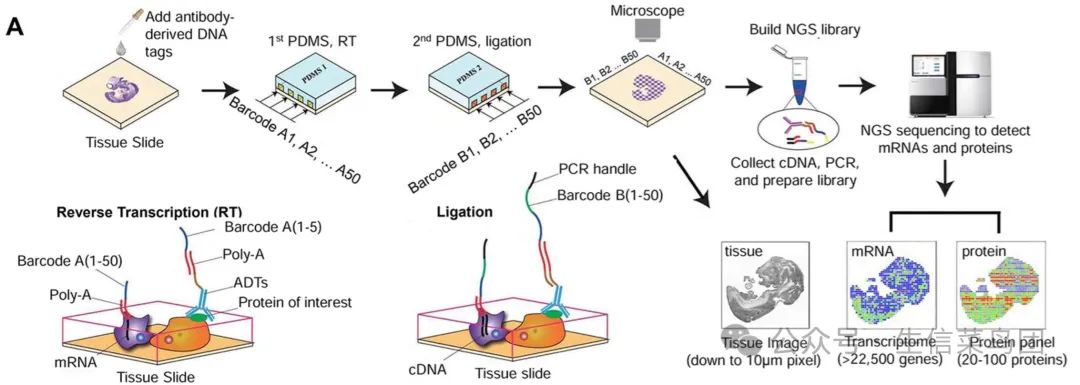
4. 基于微流控(Microfluidics)的技术
DBiT-seq(Deterministic Barcoding in Tissue for spatial omics sequencing)的核心原理是通过微流控芯片在组织表面构建确定性分布的条形码网络,将位置信息精确编码到RNA和蛋白质分子中,从而实现高分辨率的空间定位。微流控芯片由两组相互垂直的微通道组成,形成类似网格的结构。第一组通道沿水平方向排列,第二组沿垂直方向排列,两者在芯片表面交叉形成微米级的网格单元。每个通道内预先载有独特的DNA条形码探针,这些条形码由两部分组成:一组对应水平通道的位置(X轴坐标),另一组对应垂直通道的位置(Y轴坐标)。当两种条形码探针在交叉点相遇时,通过连接反应或杂交形成组合条形码,从而唯一标识每个网格单元的空间坐标。
二、评估指标
1. 分子捕获率
通过两种方式评估分子捕获率。在选定区域,要么(1)使用该区域的所有reads,要么(2)对数据进行降采样,使得不同样本具有相同数量的reads,在后续结果中将这种数据称为“降采样数据”。
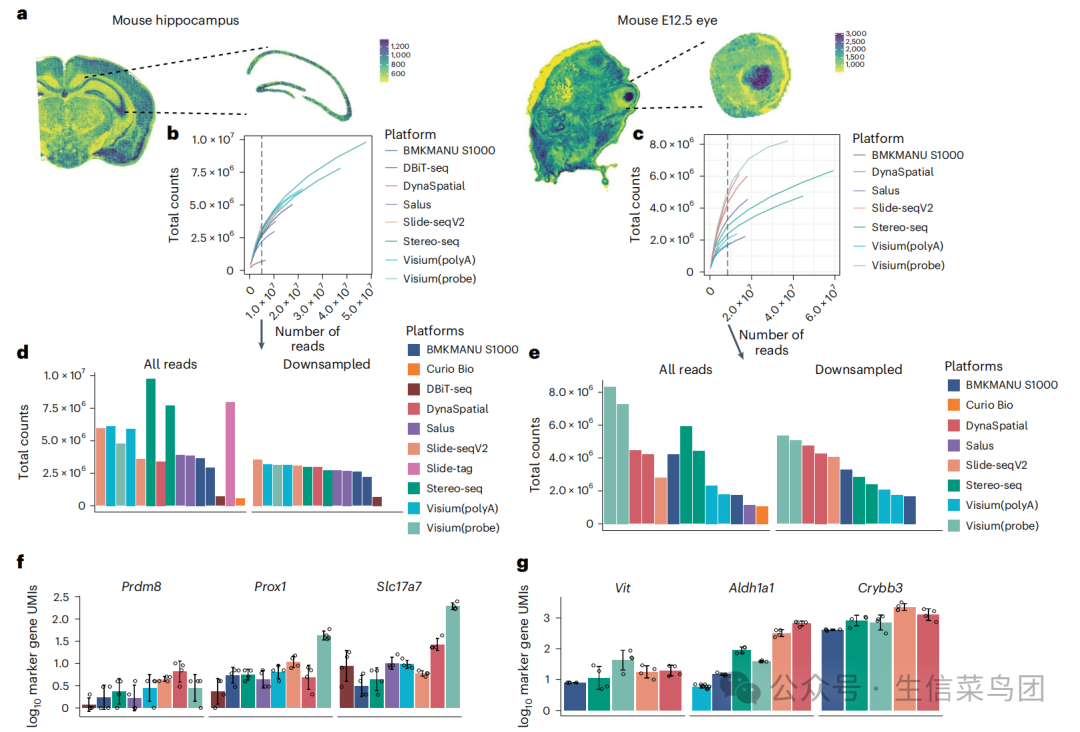
(图b,c)分别表示小鼠海马体和E12.5小鼠眼睛区域中不同检测技术得到的UMI值,横轴表示测得的reads数量,纵轴表示去除PCR重复后的有效UMI数,垂直的虚线表示降采样后的reads数。(图d,e)表示所有reads和降采样数据分别在小鼠海马体和E12.5小鼠眼睛区域的UMI数。可以看到这些测序数据都未达到饱和,并且Stereo-seq在小鼠海马体和眼睛中对同一区域的测序读数远多于其他平台。在E12.5小鼠眼睛中Visium(probe)的测序深度仅为Stereo-seq的一半左右,但仍呈现出最高的总计数,这可能是由于基于探针的方法具有更好的reads捕获效率,以及使用探针时可能存在的UMI数过度量化的影响。
作者在小鼠海马体A3区域挑选了50×50μm范围内的三个特征基因Prdm8、Prox1、Slc7a7的表达(图f),以及在E12.5小鼠眼睛区域,比较了Vit、Aldh1a1、Crybb3的表达(图g),结果显示Visium(probe),Slide-seq V2和DynaSpatial表现出高灵敏度,而Visium(polyA)灵敏度不够高。
2. 分子横向扩散
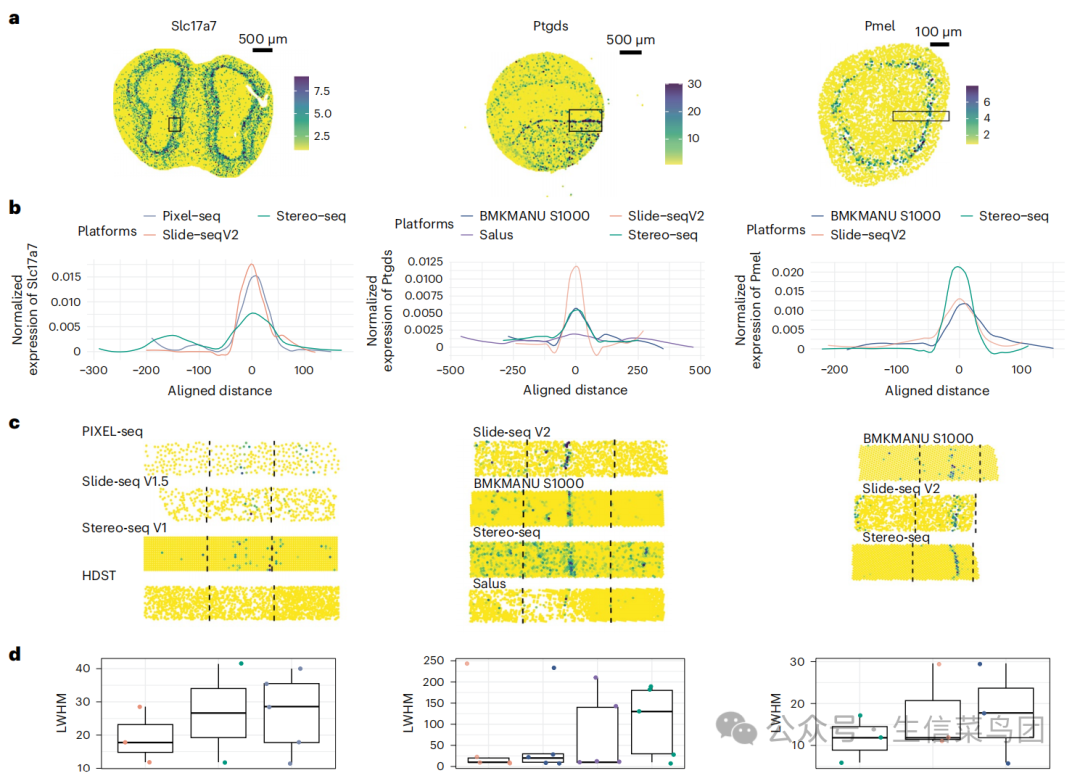
除了单位面积内的分子捕获灵敏度外,另一个关键的质量参数是mRNA检测的空间准确性。为了评估这种准确性,作者使用了两种分析方法来测量分子的横向扩散:(1)绘制特定基因在选定区域内的密度分布图;(2)量化选定区域内峰最大值一半处的左宽距离(LMWH,left width at half maximum),LWHM越小表示扩散控制越好。这些分析是基于所有reads的数据进行的。
(图a)在小鼠嗅球样本中,作者挑选了Slc7a7基因作为marker基因,因为其预期在僧帽细胞(mitral cells)和簇状细胞(tufted cells)中特异性表达,并形成独特的两个层。在小鼠脑中的marker基因为Ptgds,因为通过原位杂交确认它特异性地表达于血管细胞内的特定位置。在E12.5小鼠眼睛的marker基因为Pmel,因为它在黑色素细胞中特异性表达,这些细胞围绕着晶状体并形成圆形图案。(图b)表示归一化后的基因表达强度沿选定区域的分布,以密度图显示(面积归一化为1)。(图c)表示各技术在选定区域内的具体表达强度,黑色虚线为扩散边界。(图d)为显示各技术的LWHM值的箱线图,点表示单个样本的LWHM。
在小鼠嗅球样本中,Stereo-seq显示显著横向扩散,而Slide-seq V1.5和PIXEL-seq扩散控制较好。在小鼠脑中Stereo-seq数据存在严重扩散,且扩散问题无法通过降低测序深度解决,Slide-seq V2和BMKMANU S1000表现更优。而在E12.5小鼠眼睛中,Stereo-seq扩散控制最佳,Slide-seq V2次之。组织类型的差别和透化时间的不同会显著影响扩散模式,如Stereo-seq在眼睛中表现优异,但在大脑和嗅球中扩散较明显,未来技术发展需进一步优化扩散控制和实验条件标准化。
3. 聚类和细胞注释
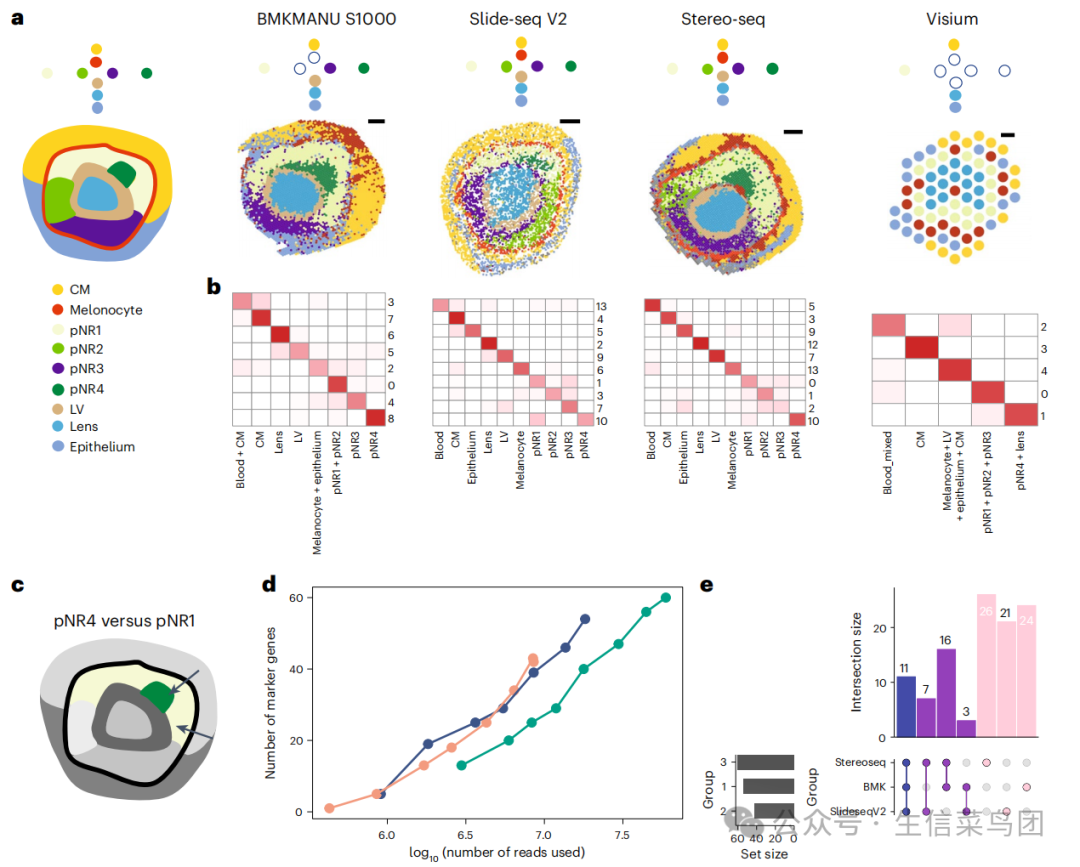
在空间转录组数据中,聚类分析与细胞注释是解析组织异质性的核心步骤。作者选择了E12.5小鼠的眼睛,这种眼睛以其独特的结构著称,即晶状体被视网膜包围,然后是黑色素细胞。
在深入比较分析之前,(图a)展示了我们对E12.5小鼠眼睛中预期观察到的细胞亚群的研究结果。在这个组织中,预期的形态结构从最内层空间展开,该空间包含晶状体(Lens)和晶状体泡(LV),这些结构被神经视网膜细胞(pNR1、pNR2、pNR3、pNR4)包围,在特定位置形成不同的亚群。神经视网膜细胞周围是黑色素细胞(Melonocyte),前侧是角膜间充质细胞(CM),而后侧则由上皮细胞(Epithelium)组成。Slide-seq V2和Stereo-seq能够很好地对每个点进行区分,进行全面的亚群注释,而BMKMANU S1000在识别黑色素细胞时遇到了挑战,这可能源于BMKMANU S1000在小鼠眼睛中具有明显的横向扩散。低分辨率的方法如Visium、DynaSpatial在确认细胞类型时受限,这主要还是因为相对低的物理分辨率。
(图b)展示了降低采样量对聚类结果的影响。图中,横轴为所有reads的聚类结果,纵轴为降采样数据的聚类结果。比较可以发现,降采样数据能够检测到几乎所有的细胞亚群,降采样对聚类结果的影响不大。
(图c)是比较神经视网膜细胞pNR1和pNR4这两种高度相似的细胞,(图d)量化地展示了三种方法用不同的测序数据量找到的marker基因的数量。随着reads的增加,marker基因的数量也随之增加,其中Slide-seq V2的方法表现出最高的敏感性。
(图e)表示各技术平台之间发现的marker基因的数目,结果显示每个技术平台发现的独特基因要比共同marker基因要多。
三、总结
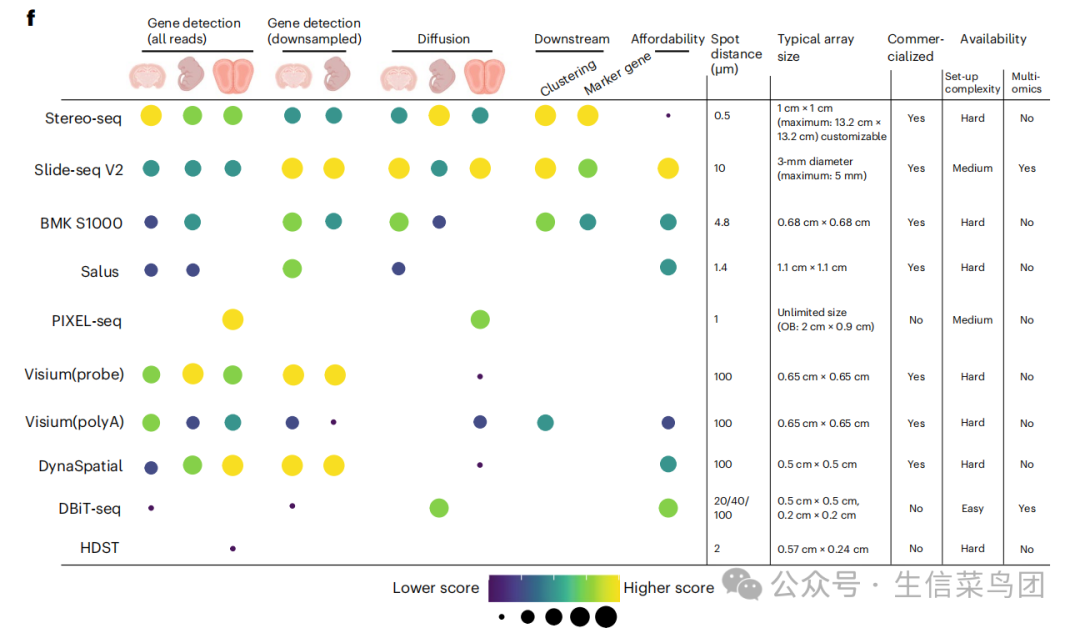
作者对各项技术从捕获效率、扩散等到下游分析的各项指标进行了总结。各项指标从左到右分别是全部测序数据检测到的基因、降采样数据检测到的基因、扩散程度、下游的聚类和marker基因、可负担性(价格)、点的距离(μm)、典型的阵列大小、是否可客户定制、建立方法的复杂度、多组学。
结果表明,空间转录组学需要更多的测序才能达到饱和水平,而本研究生成的数据远低于饱和水平。Stereo-seq、Slide-tag、Visium(probe)在原始测序深度方面表现出更好的捕获效率,而Slide-seq V2、Visium(probe)、DynaSpatial在降采样测序深度方面表现出更好的捕获效率。透化时间对扩散有显著影响,在针对每种组织类型优化透化时间后,不同技术在各种组织类型中表现出不同的扩散特征。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-05-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号