表观程序 | Nature | 多尺度足迹揭示了顺式调控元件的组织结构

表观程序 | Nature | 多尺度足迹揭示了顺式调控元件的组织结构

生信菜鸟团
发布于 2025-02-27 14:26:35
发布于 2025-02-27 14:26:35
Basic Information
- 英文标题:Multiscale footprints reveal the organization of cis-regulatory elements
- 中文标题:多尺度足迹揭示顺式调节元件的组织
- 发表日期:22 January 2025
- 文章类型:Article
- 所属期刊:Nature
- 文章作者:Yan Hu | Jason D. Buenrostro
- 文章链接:https://www.nature.com/articles/s41586-024-08443-4
Abstract
Para_01
- 顺式调节元件(CREs)控制基因表达,并且它们的结构和功能是动态变化的,反映了不同效应蛋白组成随时间的变化。
- 然而,测量全基因组上CREs处效应蛋白组织的方法有限,阻碍了将CREs结构与细胞命运和疾病中的功能联系起来的努力。
- 在这里,我们开发了PRINT,这是一种计算方法,它从多个尺度上的蛋白质大小的染色质可及性数据中识别DNA-蛋白质相互作用的足迹。
- 利用这些多尺度足迹,我们创建了seq2PRINT框架,该框架使用深度学习来精确推断转录因子和核小体结合,并解释CREs处的调控逻辑。
- 将seq2PRINT应用于来自人骨髓的单细胞染色质可及性数据,我们观察到在造血过程中围绕开路者因子的CREs的顺序建立和扩展。
- 我们进一步发现了小鼠造血干细胞中CREs结构与年龄相关的改变,包括广泛减少的核小体足迹以及新识别的Ets复合基序的获得。
- 总体而言,我们建立了一种方法,可以从染色质可及性数据中获得关于DNA结合蛋白动力学的深刻见解,并揭示分化和老化过程中调控元件的架构。
Main
Para_01
- 通过稳态、发育和疾病,细胞利用顺式调节元件(CREs)来调控基因表达。
- CREs整合了结构多样的调节蛋白的结合,这些蛋白质动态地移动以招募或驱逐协同因子,并决定了细胞的整体功能和潜力。
- 在功能基因组学中的一个主要挑战是识别所有细胞类型中这些调节蛋白的确切基因组位置和动态变化,以理解遗传网络的逻辑并破译非编码遗传变异的功能。
- 这一挑战因其复杂性和规模而显得尤为显著:在人类中,细胞使用大约2,000种转录因子(TFs)的不同组合来调节约100万个候选CREs(cCREs)的活性,这些候选CREs调控着大约30,000个基因的表达。
- 为了破解这种调控复杂性,已经在广泛的调节蛋白和细胞背景下进行了数千次染色质免疫沉淀测序(ChIP-seq)实验。
- 然而,这些努力受到限制,因为基于ChIP的方法无法扩展到测量所有细胞背景下所有调节蛋白的结合。
Para_02
- 单细胞转座酶可及染色质测序(scATAC-seq)技术已经成为一种强大且可扩展的工具,用于测量胎儿和成人组织中全部细胞多样性中的cCREs的可及性。
- 由于转录因子主要结合开放染色质,因此经常使用TF基序与可及区域的交集作为TF结合的代理。
- 为了实现更高的精度,统计方法利用染色质可及性通过量化DNA免受DNase、MNase或Tn5切割的保护来‘足迹化’cCREs上的蛋白质结合。
- 然而,足迹化方法受限于酶的序列偏差,主要关注TF尺度对象(大约20个碱基对(bp)),无法检测大量TF或不适用于单细胞方法。
- 深度学习领域的最新进展对于基因调控的各个方面研究非常有价值,允许从生物学数据中进行新解释和计算机模拟序列特征。
- 受这些进展的启发,我们寻求将DNA足迹化的精确性与深度学习的推断能力结合起来,从scATAC-seq数据中生成高基因组和细胞状态分辨率的多种调节蛋白的准确图谱。
Para_03
- 我们描述了cCREs的两步解码过程。
- 首先,我们创建了一种新的工具,命名为PRINT(用于‘通过转座进行核苷酸分辨率的蛋白质-调节元件相互作用’),该工具纠正了酶切序列偏差,并定义了cCREs的多尺度足迹表示,展示了不同大小的调节蛋白(例如,TFs和核小体)。
- 然后,我们开发了一个名为seq2PRINT的深度学习框架,它解析了cCREs中多尺度足迹的序列级组织。
- 我们发现seq2PRINT能够在bulk和scATAC-seq中实现计算上可行且精确的TF结合预测。
- 我们将seq2PRINT应用于人类骨髓细胞的scATAC-seq和RNA测序(RNA-seq)分析,并追踪了造血过程中TF和核小体的结合动态。
- 我们发现许多cCREs在分化过程中表现出调节TF的切换,这种现象并未反映在整体可及性中。
- 通过追踪分化过程中的调节变化,我们阐明了一个逐步激活红系和淋巴系cCREs的模型。
- 因为表观遗传改变,包括异常的核小体重排,是衰老的一个标志,所以我们检查了小鼠HSCs在整个生命周期中cCRE的变化。
- 我们发现了cCRE内核小体定位的全局改变,并确定了与年龄相关的TF的关键特征。
- 这些特征包括核小体相关TF(如Yy1和Nrf1)活性的降低,以及Ets和Runx家族成员在广泛共结合配置中代表的新基序的结合增加。
- 总之,这些结果表明,多尺度足迹法结合深度学习序列模型是一种强大的方法,可以预测TF结合并阐明整个基因组规模上cCREs的结构动力学。
Identification of multiscale footprints
Para_01
- 我们开发了PRINT,一种从批量或单细胞ATAC–seq数据中检测DNA结合蛋白足迹的计算方法(图1a)。
- 为了首先克服Tn5转座酶的序列偏差,这可能会显著干扰足迹检测,我们在从细菌人工染色体(BACs)去蛋白化DNA上的Tn5插入数据上训练了一个卷积神经网络(扩展数据图1a–d和补充表1)。
- 该模型在高GC含量区域的表现尤其出色,在人类基因组提取数据上的Tn5插入数据上表现相似良好(R = 0.92),并且优于ChromBPNet15的Tn5偏差校正(扩展数据图1e–k)。
- 我们提供了人类基因组和常见模式生物的预计算Tn5偏差预测,并提供了一个预训练的深度学习模型作为对该领域的资源(‘数据可用性’)。
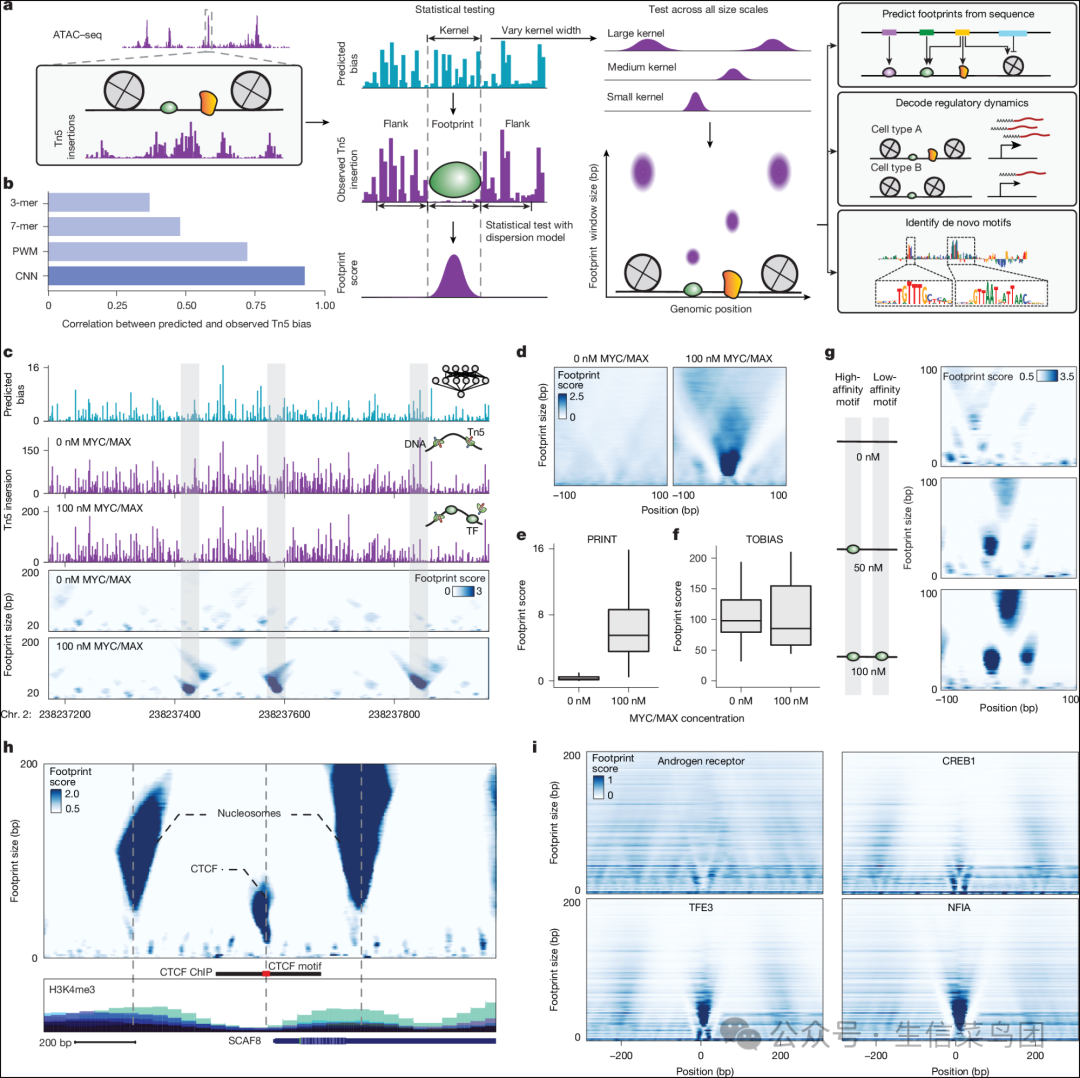
Fig. 1: Multiscale footprinting detects DNA–protein interactions across spatial scales.
- 图片说明
◉ a, 多尺度足迹工作流程概述。◉ b, 比较不同Tn5偏差校正模型性能的条形图。◉ c, 在包含0 nM(顶部)或100 nM(底部)MYC/MAX的BAC DNA上,在示例区域chr. 2:238237173–238237972处预测的Tn5偏差、观察到的Tn5插入和多尺度足迹。◉ d, 在包含0 nM或100 nM MYC/MAX的BAC DNA上,在MYC/MAX基序位点上的聚合多尺度足迹。◉ e, f, 在包含0 nM或100 nM MYC/MAX的MYC/MAX基序位点上的足迹评分箱线图,显示PRINT(e,位置数= 275)和TOBIAS18(f,位置数= 158)的结果。◉ 盒子显示第一、第二和第三四分位数,须显示最远点落在第一四分位数减去1.5倍四分位距(IQR)或第三四分位数加上1.5倍IQR范围内。◉ g, 在包含0 nM(顶部),50 nM(中间)和100 nM(底部)MYC/MAX的两个相邻MYC/MAX基序位点上的多尺度足迹。◉ h, 在HepG2细胞中的cCRE区域chr. 6:154732971–154733770内的多尺度足迹。下层轨道是叠加的ENCODE组蛋白ChIP信号。◉ i, 包括AR、CREB1、TFE3和NFIA在内的示例转录因子的聚合多尺度足迹。CNN,卷积神经网络。
Para_02
- PRINT 在蛋白质大小的不同尺度上以高灵敏度和特异性识别足迹。
- 我们开发了一种统计方法,量化观察到的 Tn5 插入相对于给定位置估计的背景分散的显著耗竭,从而得出足迹评分(方法部分)。
- 这种方法将去蛋白 DNA 上假阳性检测的数量减少了一个数量级,与之前的足迹法相比18(扩展数据图 1l-v)。
- 受先前使用 MNase19 或 ATAC-seq5,20,21 片段大小推断不同大小的 DNA 结合蛋白的方法启发,我们接下来计算了跨越 4-200 bp 窗口大小的足迹评分。
- 我们在体外通过去蛋白 DNA 与纯化的 MYC/MAX 或 CEBPA 孵育验证了该方法。
- 仅在存在纯化 TF 的情况下,在 TF 基序位点检测到了强足迹,而背景信号非常低,而一个已建立的 ATAC-seq 足迹法18没有检测到前景和背景之间的任何区别(图 1c-f 和扩展数据图 2a-d)。
- 此外,我们发现在较高浓度(100 与 50 nM)下 MYC/MAX 在低亲和力位点上的足迹增加,这表明足迹评分对给定点的 TF 占用率敏感(图 1g)。
Para_03
- 我们发现PRINT可以检测哺乳动物细胞中的足迹。
- 我们观察到与核小体和特定转录因子相对应的独特足迹模式(图1h-i)。
- 转录因子结合模式可以被聚类成四个代表性类别(扩展数据图2e)。
- 与先前使用DNase I的研究一致14,我们发现转录因子之间的足迹强度各不相同,包括一些没有留下可检测足迹的转录因子,这可能是由于它们在DNA上的结合较弱或短暂。
- 抑制性转录因子的足迹也可以被检测到(扩展数据图2f)。
- 我们通过与外切酶染色质免疫沉淀(ChIP-exo)的数据进行基准测试来验证足迹,在转录因子结合位点上发现了符合,并且发现了ChIP-exo的可能假阴性结果(扩展数据图2g-h)。
- 综上所述,我们的结果显示,使用PRINT进行多尺度足迹分析可以稳健地检测许多不同的DNA结合蛋白。
A DNA sequence model for footprints
Para_01
- 我们接下来试图使用多尺度足迹来预测特定蛋白质与DNA的结合。
- 我们设计了模型来预测转录因子和核小体的结合(图2a,方法和补充说明)。
- 核小体模型使用多尺度足迹作为输入来预测由核小体化学映射数据映射的核小体峰;我们发现该模型的表现优于之前的工作(扩展数据图3a-f)。
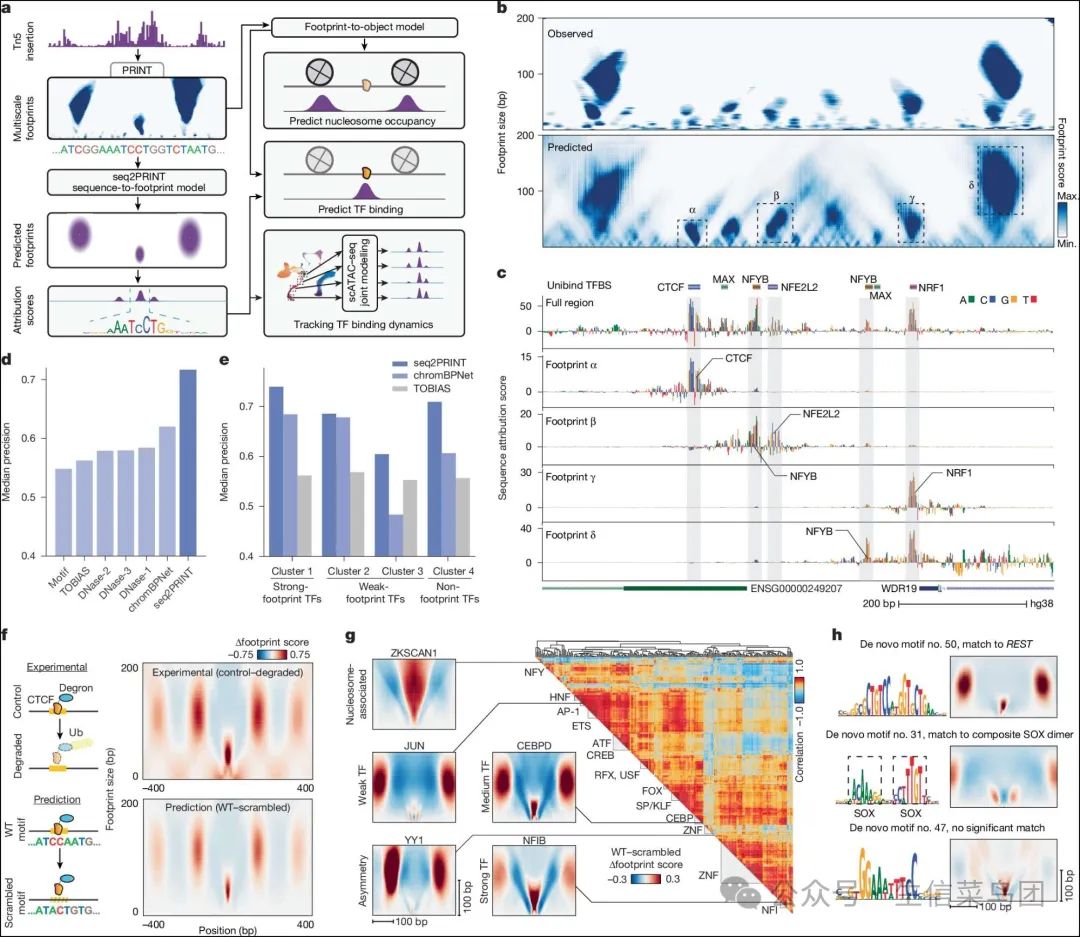
Fig. 2: Decoding the genomic syntax of cCRE organization.
- 图片说明
◉ a, 足迹到对象预测和seq2PRINT模型及其应用的示意图。◉ b, 示例区域chr. 4:39181940–39182739中的观察到的(顶部)和预测的多尺度足迹(底部)。◉ c, b图中所示区域的seq2PRINT序列归因分数,显示了根据整个区域内的多尺度足迹(第1条轨迹)或特定足迹(第2-5条轨迹)计算的归因分数。◉ d, 条形图显示不同方法预测TF结合的中位精度。◉ e, 条形图显示seq2PRINT、ChromBPNet和TOBIAS对于不同TF簇(如扩展数据图2e中定义的)预测TF结合的中位精度。◉ f, 左侧,实验性去除degron标记的CTCF和计算机模拟破坏CTCF结合位点的示意图。右侧,实验性去除CTCF后CTCF基序位点处多尺度足迹的变化,以及计算机模拟CTCF基序破坏后seq2PRINT预测的多尺度足迹变化。◉ g, 左侧,计算机模拟去除示例TF基序位点后seq2PRINT预测的多尺度足迹变化。右侧,基于计算机模拟基序去除后seq2PRINT预测的变化对TF进行聚类的热图。◉ h, 通过从seq2PRINT预测位点的序列归因分数(TF-MoDISco)识别出的代表性的新基序,以及相应的计算机模拟基序去除后预测的多尺度足迹变化(颜色标度同g图)。最大值,最大;最小值,最小。
Para_02
- 基于近期深度学习的进展,我们创建了一个模型,该模型使用DNA序列来预测多尺度足迹(seq2PRINT;图2a和扩展数据图4a)。
- 该模型仅以局部DNA序列为输入就能预测核小体和转录因子足迹(图2b)。
- 我们观察到,在来自HepG2细胞的ATAC-seq数据中,预测的和观测的多尺度足迹之间的总体相关性为0.75,并且这种相关性在读取深度的子采样中保持稳健(扩展数据图4b)。
Para_03
- 我们提取了seq2PRINT学习到的关键序列特征,并发现由此产生的基于碱基的DNA序列归因评分能够解析cCRE内的TF结合架构。
- 在一个例子位点中,相对于整个cCRE计算的归因评分突出了跨越该区域的TF基序位置重叠的短序列,而针对特定足迹对象计算的评分则突出了特定的基序(图2b,c)。
- 对于足迹α和γ,序列模型识别出该足迹下的基序,而对于足迹β,不仅检测到了足迹下的NFE2L2基序,还检测到了一个邻近的NFYB基序,表明附近TF之间的潜在结合协调。
- 足迹δ可能代表一个核小体,并且由附近的TF NRF1和NFYB基序预测,显示了更长范围的依赖关系,揭示了与核小体定位最相关的因素。
- 后两个例子进一步表明,某些缺乏强烈足迹的TF(例如,NFYB)可以通过seq2PRINT被检测到,这可能是由于对邻近元件的影响,而且这种方法可以用于建模cCRE内DNA结合蛋白之间的相互作用。
Para_04
- seq2PRINT可以用于预测TFs的全基因组结合。我们使用seq2PRINT的序列归因评分来生成一个训练用于预测ChIP–seq数据的TF结合评分(方法)。
- 该TF结合评分能够高精度地预测TF结合,并且优于先前的方法15,18(图2d)。
- 我们发现,即使对于其他方法表现特别差的弱结合或没有直接足迹的TF,我们也能够进行预测(图2e,扩展数据图2e和4c–f以及补充表2)。
- 为了研究模型如何检测这些TF,我们通过打乱它们的基序序列来模拟TF结合丢失的效果。
- 模型预测了附近足迹的变化,包括核小体等不同尺度上的变化,这些变化与实验确定的足迹变化高度相关,包括degron诱导的CTCF耗竭4(综合R=0.93,每个位置的中位数R=0.66;图2f)。
- 类似的实验结果也出现在地塞米松诱导的糖皮质激素受体重定位和DNA结合24,以及IFN诱导的Stat2结合到DNA25(扩展数据图4g–j)。
- 我们将这种方法广泛应用于识别对TF相邻核小体具有强烈影响的TF(例如JUN和YY1),或者在同一位置的TF(例如ZKSCAN1)(图2g)。
- 预测的足迹评分变化在TF家族之间高度相似,这种相似性在很大程度上独立于结合位点的相似性(扩展数据图4k)。
Para_05
- seq2PRINT 属性评分识别出可预测足迹的关键 DNA 序列模式,使从头鉴定基序成为可能。
- 利用训练好的模型,我们聚类和对齐了局部序列属性评分,并鉴定了 106 个从头基序。
- 这些从头基序以无偏的方式恢复了已知基序,以及如 SOX 二聚体等复合基序(图 2h)。
- 几个从头基序与强烈的转录因子或核小体足迹相关,尽管它们与已知基序数据库没有显著匹配(图 2h,底部)。
- 我们在补充数据 1 和 2 中提供了在这项工作中发现的所有从头基序及其关联的多尺度足迹的完整列表。
cCREs restructure across haematopoiesis
Para_01
- 我们使用同时进行的高通量ATAC和RNA表达测序(SHARE–seq)27来生成来自七名人骨髓单核细胞供体的874,480个骨髓单核细胞的联合scATAC–seq和scRNA–seq数据集(图3a和扩展数据图5a–g)。
- 为了实现足迹化,我们将单个细胞合并成1,000个伪整体,代表所有主要细胞类型和发展转换(扩展数据图5h–m和方法部分)。
- 在应用深度学习序列模型时面临的一个核心挑战是计算强度随着细胞类型或条件数量的增长而难以扩展。
- 为了解决这一挑战,我们在代表22亿读取的所有数据上训练了一个通用模型,并使用大规模模型的低秩适应(LoRA)对每个伪整体的序列模型进行微调28,与为每个伪整体单独训练模型相比,这种方法大约提高了80倍的速度,并且预测准确性也有所提高(扩展数据图6a–c)。
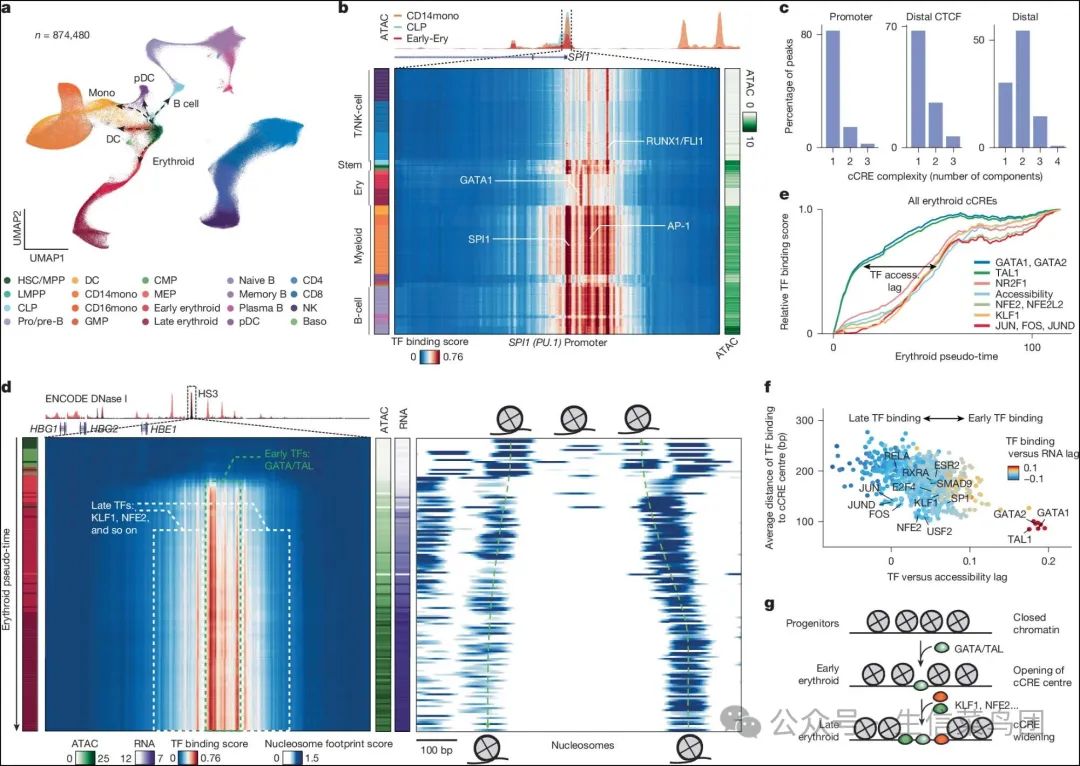
Fig. 3: Emerging intra-cCRE dynamics in human haematopoiesis.
- 图片说明
◉ 均匀流形逼近和投影(UMAP)的人类骨髓SHARE–seq数据集。◉ 顶部,围绕突出显示的SPI1启动子区域chr. 11:47378111–47378911的ATAC–seq信号。◉ 底部,在同一SPI1启动子区域内1,000个伪批量中由seq2PRINT得出的TF结合评分。◉ 每行代表一个伪批量,每列代表单个碱基位置。◉ 左侧颜色图例与a相同;右侧颜色图例,每个伪批量中启动子区域的可访问性。◉ 所有cCREs中具有给定复杂度的TF结合模式的百分比。◉ 各个cCRE结合模式被分解为主成分,复杂度定义为解释98%方差所需的组件数量。◉ 左侧,在染色体控制区域HS3增强子区域chr. 11:5284362-5285162内,在红系分化过程中由seq2PRINT得出的TF结合评分。◉ 行表示按伪时间排序的伪批量。◉ 颜色图例与a相同。◉ 中间,彩色条表示同一区域的ATAC–seq信号以及HBB的RNA水平。◉ 右侧,同一区域和伪批量中的核小体足迹评分(100 bp比例尺)。◉ 线条图显示了红系cCREs中代表性TFs的TF结合评分随红系分化伪时间的变化。◉ 散点图比较了TF结合的时间和TF结合位点相对于cCRE中心的位置。◉ X轴:TF结合与cCRE可访问性获得之间的时间滞后;Y轴:TF结合位点到cCRE中心的平均距离。◉ 示意图展示了分化过程中TF的顺序结合和cCRE的扩大。◉ HSC/MPP,造血干细胞/多能祖细胞;LMPP,淋巴样倾向多能祖细胞;CLP,共同淋巴样祖细胞;pro/pre-B,前体B细胞祖细胞和前体B细胞;DC,树突状细胞;CD14mono,CD14单核细胞;CD16mono,CD16单核细胞;GMP,粒细胞-单核细胞祖细胞;CMP,共同髓样祖细胞;MEP,巨核细胞-红细胞祖细胞;early-Ery,早期红细胞;late-Ery,晚期红细胞;Naive B,幼稚B细胞;Memory B,记忆B细胞;Plasma B,浆细胞B细胞;pDC,浆细胞样树突状细胞;CD4,CD4 T细胞;CD8,CD8 T细胞;NK,自然杀伤细胞;Baso,嗜碱性粒细胞。
Para_02
- 转录因子结合预测显示了在同一细胞类型中不同组的TF结合在相同的cCRE上。在SPI1(PU.1)启动子处,seq2PRINT结合评分在髓样细胞中的SPI1和AP-1位点上较高,而在红系细胞中仅预测出强烈的GATA1结合,这与这些TF已知的调控关系一致29(图3b)。值得注意的是,这些不同的TF结合模式以及在其他位点上的结合模式不能仅通过测量启动子的整体可及性来区分(扩展数据图6d-g)。我们使用主成分分析量化了基因组每个cCRE上的TF结合模式的复杂性,并发现复杂的cCRE(多于一个成分)相对于启动子而言在远端cCRE上高度富集(69.8对17.2%;图3c),突显了增强子在细胞类型特异性利用方面的特点。
Para_03
- 分析沿红系分化轨迹的TF结合显示,cCRE的建立是逐步发生的。这可以通过血红蛋白基因座控制区内的HS3增强子来说明。
- 在造血干细胞和共同髓样祖细胞中,核小体是无相位的,并且预测TF结合较低。
- 随着细胞沿着红系发育进程前进,按伪时间顺序排列,并且HBB(β-血红蛋白)表达增加,核小体印迹首先在cCRE边缘处形成相位,并在GATA/TAL基序位点具有强烈的TF结合评分。
- 然后,cCRE逐渐扩大,在边缘增加了KLF1/NFE2因子的结合(图3d和扩展数据图6h)。
- HBB启动子表现出相同的逐步TF结合模式,并且该位置的PRINT/seq2PRINT结合预测与先前发表的大规模平行报告测定数据很好地对应(扩展数据图6i–m)。
Para_04
- 我们发现了这种cCRE模式从一个中心TF向外扩展到整个红细胞相关cCRE的基因组。
- GATA和TAL因子的结合评分在红细胞伪时间早期增加,
- 而显著的整体cCRE开放以及NFE2、KLF1、NR2F1和AP-1因子的结合则在分化后期发生(图3e和扩展数据图6n)。
- 引人注目的是,预测在可及性增加之前结合并靶向基因表达的TF也更靠近cCRE中心结合,
- 而晚期结合的TF基序定位在侧翼区域(图3f、g和补充表3)。
- 这些观察结果在很大程度上独立于cCRE的复杂性(扩展数据图6o-r)。
- 这一观察结果与先前研究表明的cCRE内TF基序排列的典型性和cCRE边缘染色质重塑器的富集一致5,33,34。
- 我们在B细胞分化轨迹上重复了这一分析,并发现具有不同中心和侧翼TF的cCRE的相同顺序建立(扩展数据图6s)。
- 我们的研究结果将体内分化过程中的cCRE重构和TF结合联系起来,表明核心结合和侧翼结合的TF可能在不同程度上对cCRE的建立、扩展和基因表达做出贡献。
De novo motifs characterize ageing HSCs
Para_01
- 我们接下来试图使用seq2PRINT来研究衰老过程中cCRE组织的变化。
- 生物衰老是一个多因素过程,影响广泛的组织的生理机能,包括显著的功能和造血干细胞(HSCs)增殖的变化。
- 由于先前的研究表明衰老伴随着广泛的表观遗传变化,
- 我们假设seq2PRINT将非常适合检测不同年龄阶段的TF活性和cCRE结构差异。
- 我们通过荧光激活细胞分选(FACS)从年轻(11周大,n = 10)和年老(24个月大,n = 5)雄性小鼠的骨髓中分离出造血祖细胞(Lin−)和HSCs(Lin−Sca1+cKit+CD48−CD150+),
- 并使用10x Multiome平台对48,225个细胞进行了联合ATAC-RNA分析,涵盖了14,640个HSCs和33,585个造血祖细胞(图4a、b及扩展数据图7a-g)。
- 来自老年和年轻小鼠的HSCs分别聚类,并与之前报道的衰老特征和标记基因相匹配(图4c、d,扩展数据图8a-e和补充表4)。
- 为了进一步探索HSC异质性,我们在HSCs中鉴定了100个代表性细胞状态,以生成伪整体。
- 我们通过收集先前描述的涵盖谱系偏差和衰老的基因集以及从我们的数据中学到的基因程序来识别HSC状态(方法)。
- 使用这些基因集对HSC伪整体进行评分和聚类,确定了五种由年龄和谱系潜力区分的HSC亚群(图4e、f和扩展数据图8f-m)。
- 这五种HSC细胞状态代表了与先前发现一致的主要类别,并且可以进一步划分,表明了与年龄相关的变异的其他轴(例如,氧化磷酸化调控,未折叠蛋白反应)可能与老年人免疫系统的发病机制相关(补充表5)。
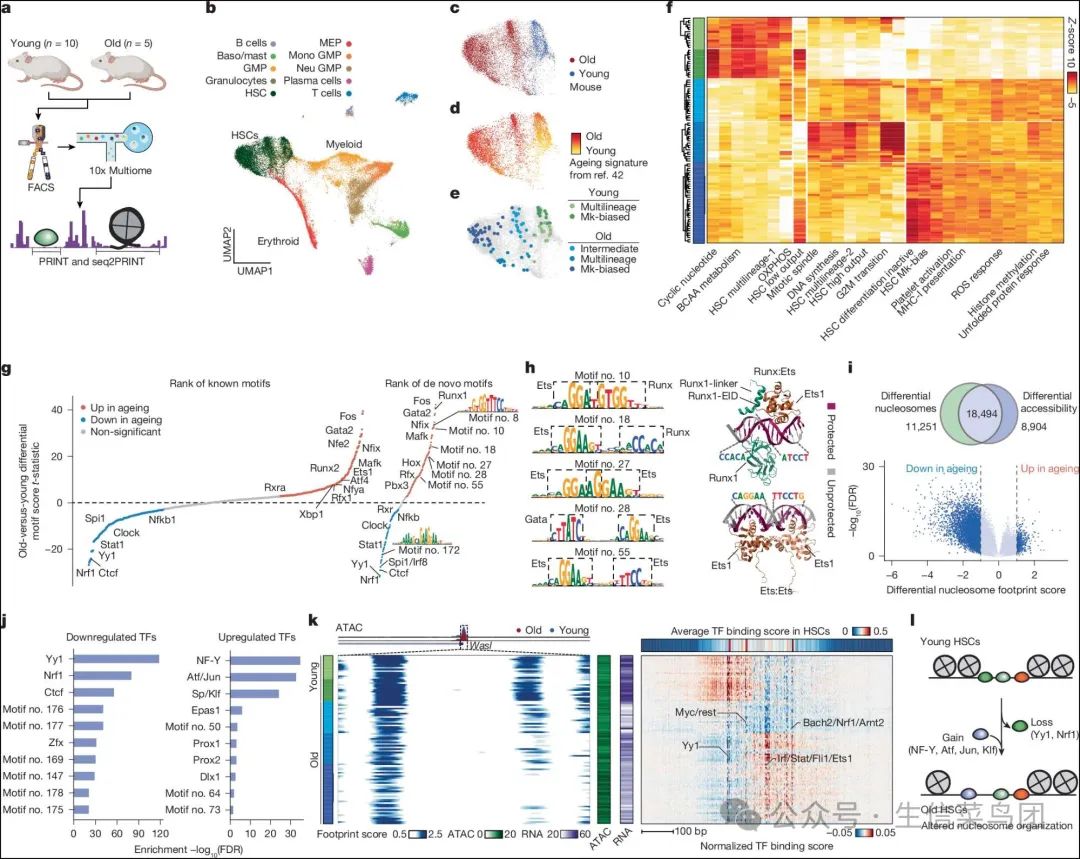
Fig. 4: Intra-cCRE dynamics in haematopoietic ageing.
- 图片说明
◉ a, 数据生成和分析的示意图。◉ b, HSC和祖细胞的UMAP图。◉ c, 展示供体小鼠年龄的HSC的UMAP图。◉ d, 展示从参考文献42中获得的老化基因特征活性的UMAP图。◉ e, 展示五个主要HSC亚群的UMAP图。◉ f, 使用基因程序表达来将100个HSC伪批量聚类成五个主要亚群的热图。色标表示每个伪批量的HSC亚群标签,如e所示。◉ g, 已知(左侧,来自cisBP)和新发现(右侧,由seq2PRINT衍生)TF基序按老年与年轻差异染色质变异评分测试t统计量(双尾)排名。显著的cisBP匹配没有的顶级基序被突出显示。◉ h, Ets同/异二聚体的示例复合新基序,还展示了PDB结构的二聚体Runx:Ets(PDB-4L0Z)和Ets:Ets(PDB-2NNY)。溶剂不可及核苷酸碱基被突出显示为紫色,并显示核心Runx和Ets基序。◉ i, 与年龄相关的核小体变化。顶部,展示在老化过程中具有差异核小体和差异可及性的cCRE重叠的维恩图。底部,老化过程中差异核小体变化的火山图,用密度伪彩色。◉ j, 在老化过程中丢失的核小体足迹处的TF基序富集(足迹得分的绝对差异大于1且错误发现率(FDR)小于0.01)。上调节(右侧)或下调节(左侧)的前十个富集基序。◉ k, 在老年和年轻HSC中chr. 6:24664695–24665494位置Wasl启动子的组织。左侧,核小体足迹(100 bp比例尺);行代表单个HSC伪批量,列代表单个bp位置。左侧色标表示HSC亚群,如e,f所示;中间色条表示伪批量中的染色质可及性和Wasl RNA水平;右侧,顶部色条表示跨伪批量的平均seq2PRINT预测的TF结合评分。热图显示每个伪批量中的TF结合评分,在通过减去列均值进行居中后。◉ l, 与年龄相关的cCRE重组的示意图。◉ a中的插图使用BioRender创建(https://biorender.com)。
Para_02
- 我们发现染色质可及性区域(cCREs)在衰老过程中经历了转录因子(TF)结合的广泛变化。
- 我们在所有HSC伪群体上应用了seq2PRINT,并比较了年轻和年老的HSC,发现NF-I、Runx、Ets、Gata和AP-1(例如Fos、Jun)的活性显著增加(图4g和补充表6)。
- 考虑到将所有年轻和年老的HSC进行比较可能会受到衰老过程中HSC亚群组成变化的影响,我们分别使用仅偏向巨核细胞(Mk)的亚群和多系亚群进行了这一分析。
- 特定亚群的比较总体上得到了相似的结果,表明大多数变化在亚群之间是共享的(扩展数据图9a-d)。
- 然而,一组Ets基序,特别是Spi1/Spib/Spic和Elf,在多系亚群中显示出与年龄相关的下调,这表明随着衰老,不同亚群中的转录因子发生了特定的变化(扩展数据图9c-d)。
Para_03
- 为了全面且不带偏见地检查seq2PRINT学习到的序列基序,我们识别了具有差异活性的新基序(图4g、扩展数据图9e和补充表6),包括衰老过程中许多富含CG的新基序的丢失,这可能与DNA结合因子对多种背景下衰老特征性甲基化变化的识别有关(扩展数据图9f)。
- 我们发现,在包含Ets同源或异源二聚体以及Gata、AP-1和Runx基序的复合基序中的活性有所增加(图4g-h和扩展数据图9g-h)。
- 此外,我们发现Ets/Runx复合基序在老化的多谱系HSC中特别丰富(扩展数据图9h-i)。
- 支持这一发现,先前的研究提出了Ets/Runx共现对HSC维持和髓样命运的作用(参考文献43)。
- 从机制上讲,Ets对DNA的结合受到自抑制结构域的负调控,该结构域在与Runx或第二个Ets家族转录因子共同结合后被释放,并且已在两个实验确定的结构(PDB-4L0Z和PDB-2NNY;图4h)中显示了物理相互作用(参考文献44,45)。
- 为了测试seq2PRINT预测的Ets复合基序是否可以代表类似的直接相互作用,我们使用AlphaFold3来预测结构,发现所有由seq2PRINT识别的Runx/Ets配置均显示出相似的物理相互作用,无论方向如何。
- 支持这些预测的有效性,基于已知Ets/Ets和Ets/Runx基序配置的AlphaFold3结构与实验测量的结构高度一致(第10个基序的预测结构与PDB-4L0Z之间的均方根偏差为0.825埃),并且每个转录因子界面处的溶剂不可及碱基匹配正确的核心基序(图4h、扩展数据图9j、补充说明和补充表7)。
- 因此,seq2PRINT揭示了衰老过程中TFs的重布线,通过新基序分析结合AlphaFold3,预测了一种广泛多样的Ets和Runx共结合排列,这些排列与衰老和HSC自我更新有关。
- 我们还提供了伪群体中的TF得分,以便探索与基因程序相关的TFs(补充表5)。
Para_04
- 在多个系统中,各种研究描述了与衰老和早衰相关的核小体的全局丢失,尽管关于这种丢失是全局性的还是局限于特定的TF相关位点仍存在争议。
- 为了进一步探索HSC表观遗传的衰退,我们使用PRINT测量了衰老过程中cCREs的核小体占用率。
- 我们观察到,在老年HSCs的cCREs中普遍存在核小体足迹的丢失(图4i和扩展数据图9k–q)。
- 我们注意到,38%具有不同核小体足迹的cCREs没有显示出显著的可及性变化,这突显了足迹分析在解析变化中的作用。
- 核小体足迹的减少并不一定意味着整个基因组中核小体数量的减少,因为这也可能是由于核小体定位/相位的丢失,而分析主要限于cCREs。
- 为了确定这些位点是否特异性地改变了特定的TFs,我们使用seq2PRINT基序,并发现老化下调的TFs如Yy1、Nrf1和Ctcf的富集,以及上调的TFs如Nfyb、Atf、Jun和Klf的增加(图4j,k和扩展数据图9m,n,p,q和10a–f)。
- 值得注意的是,Yy1、Nrf1和Ctcf也被预测在多种细胞系中调节核小体的位置(图2c,g)。
- 先前的研究已经表明,Yy1是HSC自我更新和静止的调节因子。
- 总体而言,我们在衰老过程中发现了核小体足迹的广泛改变和TF结合的重布线,这表明衰老过程中cCRE结构的失调(图4l)。
Discussion
Para_01
- 我们的结果显示了在细胞分化和衰老过程中,转录因子结合和核小体重定位在cCREs上的复杂动态。
- 先前的足迹研究显示,转录因子结合主要由cCREs的整体开启或关闭决定,而不是由相同cCRE内的转录因子差异结合决定。
- 相比之下,我们这项研究表明,cCREs在不同细胞类型中被不同的转录因子集合占据。
- 这通过上述SPI1/PU.1和Wasl启动子分析得到了体现(图3b和图4k),尽管总体可及性水平相似,但观察到核小体和转录因子的多种配置,展示了标准染色质可及性分析所遗漏的基因调控机制。
- 直接支持这一模型的研究通过ChIP–seq映射转录因子结合报告,转录因子在发育过程中会发生切换。
- 沿着造血分化的连续轨迹,我们发现cCREs在中心转录因子周围逐步扩大,而侧翼转录因子在发育后期阶段结合,表明增强子的建立是一个模拟过程,而非数字过程。
Para_02
- PRINT 更广泛地生成了给定细胞群体中所有DNA结合蛋白的图像。
- 使用seq2PRINT建模足迹可以推断TF结合,而不论其直接足迹强度如何,能够从头识别TF基序和协同结合,并表明TF具有重塑或稳定相邻核小体等专门功能。
- 这些属性使seq2PRINT与ChromBPNet15形成对比,后者被调整用于预测可及性谱图而不是解释足迹背后的序列特征。
- 通过使用LoRA28,我们减少了足迹预测的计算负担,使我们能够将seq2PRINT扩展到单细胞表观基因组学数据。
- 我们预计这种方法将使连接高分辨率足迹分析与多样化的表观基因组数据类型(如基因组结构和局部基因表达)的方法成为可能。
- 同样,识别TF结合并将足迹归因于特定的碱基对分辨率序列也可能赋予先前被峰值分析所掩盖的致病遗传变异的新功能。
Para_03
- 这些方法存在局限性。我们的方法没有包含转置片段长度,这可能成为分析的一个附加特征。
- 此外,由于深度学习需要多个实例来识别模式,罕见或非规范的结合事件可能会被忽略。
- 另外,scATAC-seq 的足迹分析需要伪批量处理以获得足够的 Tn5 插入事件来识别足迹。
- 我们设想 PRINT 可能通过其他方法得到补充,例如基于甲基转移酶的单分子足迹分析和 DNA 序列诱变测定,以进一步分析特定 cCREs 的结构和功能。
- 然而,由于 ATAC-seq 已被广泛采用并在单细胞检测中广泛应用,seq2PRINT 可能独特地使回顾性和前瞻性研究成为可能,这些研究可以跨大量健康和患病的人体组织绘制足迹。
- 综上所述,我们的方法从一维染色质可及性数据中提取丰富的多维特征空间,以揭示跨越高基因组和细胞状态分辨率的 cCREs 的动态结构。
Methods
Experimental methods
实验方法
Cell culture
细胞培养
Para_01
- HepG2细胞(ATCC HB-8065;通过短串联重复谱型分析进行鉴定,并由ATCC进行支原体检测)在DMEM(目录号11965092,ThermoFisher)中培养,其中添加了10%胎牛血清(FBS)和1%青霉素/链霉素。
- 细胞在含有5%二氧化碳的37°C环境中孵育,并保持在对数生长期,随后使用TrypLE Express(目录号12604013,ThermoFisher)进行消化以制备单细胞悬液。
BMMC sample processing
BMMC样本处理
Para_01
- 冷冻的人类骨髓单核细胞(BMMCs,AllCells;商业捐赠者细胞实验在获得哈佛机构审查委员会批准后进行)在37°C的水浴中解冻1分钟,并转移到一个15毫升的离心管中。
- 接下来,向细胞中逐滴加入10毫升预热的DMEM和10%胎牛血清,然后在室温下以400g离心3分钟。
- 去除上清液后,用含有0.04%牛血清白蛋白(BSA)的0.5毫升PBS洗涤细胞两次。
- 为了去除中性粒细胞,将细胞悬浮在含有0.2% BSA的100微升冷DPBS中,并加入10微升人TrueStain FcX(BioLegend,目录号422302),并在冰上孵育10分钟以减少非特异性标记。
- 随后,在冰上再孵育30分钟,加入0.5微升生物素抗人CD15抗体(BioLegend,目录号301913)。
- 免疫染色后,向样品中加入25微升MyOne T1磁珠,在室温下孵育5分钟,以捕获中性粒细胞。
- 然后我们向样品中加入900微升含有0.2% BSA的DPBS进行稀释。
- 将样品置于磁铁上孵育3分钟,然后在样品仍位于磁铁上时转移1毫升到一个新的试管。
- 细胞随后准备好进行固定和SHARE-seq实验。
Fixation
注视
Para_01
- 细胞以300g离心5分钟,然后在PBSI中重悬至每毫升1百万个细胞。
- 通过加入甲醛(目录号:28906,ThermoFisher)至终浓度为1%,将细胞固定,并在室温下孵育5分钟。
- 通过加入56.1微升2.5M甘氨酸,50微升1M pH 8.0的Tris-HCl和13.3微升7.5%的BSA于冰上停止固定。
- 样品在室温下孵育5分钟后,以500g离心5分钟去除上清液。
- 所有离心均在摆动桶离心机上进行。
- 细胞沉淀物用1毫升PBSI洗涤两次,在每次洗涤之间以500g离心5分钟。
- 细胞重悬于含有0.1单位每微升Enzymatics RNase抑制剂的PBS中,并分装用于转座反应。
SHARE–seq
SHARE–seq
Para_01
- 固定后,按照之前描述的方法进行了SHARE–seq实验,但做了一些修改。为了提高转座效率,使用了预组装的Tn5(seqWell,Tagify(TM) SHARE-seq试剂)。为了提高RNA捕获效率,在逆转录前向转录本中添加了poly-A。具体操作是将转座后的细胞(60 μl)与240 μl的poly(A)混合液(最终浓度为1×Maxima RT缓冲液,0.25 U μl−1 Enzymatics RNase抑制剂,0.25 U μl−1 SUPERase RI,0.018 U μl−1大肠杆菌poly-A酶(New England Biolabs目录号M0276L)和1 mM核糖核酸ATP)混合。然后将样品分装至每个PCR管50 μl,并在37 °C下孵育15分钟。
- ,
Quantification and sequencing
量化和排序
Para_01
- 这两个文库分别使用KAPA文库定量试剂盒进行了定量,并合并用于测序。
- 单细胞文库使用Nova-seq平台(Illumina)进行测序,使用了200个循环的试剂盒(读取1,50个循环;索引1,99个循环;索引2,8个循环;读取2,50个循环)。
- 批量文库使用Nova-seq平台(Illumina)进行测序,使用了100个循环的试剂盒(读取1,50个循环;索引1,8个循环;索引2,8个循环;读取2,50个循环)。
SHARE–seq data preprocessing
SHARE-seq数据预处理
Para_01
- SHARE–seq 数据使用了 SHARE-seqV2 对齐管道(https://github.com/masai1116/SHARE-seq-alignmentV2/)进行处理,并与 hg38 进行比对。
- 使用 MACS2 峰值调用器(v.2.2.9.1)对单个样本中的开放染色质区域峰值进行了调用,参数如下:--nomodel –nolambda –keep-dup -call-summits。
- 将所有样本的峰值合并,并过滤掉与 ENCODE 黑名单区域重叠的峰值(http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg38-human/hg38.blacklist.bed.gz)。
- 将峰值峰顶两侧各延伸 150 bp,并定义为可访问区域(用于足迹分析时,这些峰值稍后被调整为宽 1,000 bp)。
- 使用 chromVAR(v.1.24.0)计算峰值中的片段计数和 TF 得分。
- 删除了低于 30% 的读取数位于峰值内的细胞条形码(峰值内读取分数,FRiP),或低于 250 个独特片段。
- 然后,将对齐的读取与峰值窗口区域相交,生成一个矩阵,其中包含峰值中的染色质可及性计数(行)和细胞(列)。
- 为了检查细胞身份,使用了 cisTopic(50 个主题)进行降维,随后进行 Louvain 聚类。
- 祖细胞群体被亚聚类以获得更精细的细胞身份。
- 通过 UMAP 将数据投影到二维空间。
- 使用 Seurat v.3(v.5.0.3)按总 UMI 计数缩放数字基因表达矩阵,乘以转录物数量的平均值,并且值进行了对数转换。
Generation of BAC naked DNA data
BAC裸DNA数据的生成
Para_01
- 我们基于与手动选择的一组关键转录因子和分化相关基因的重叠选择了25个染色质区域。
- 细菌人工染色体(BAC)克隆(BACPAC资源)在裂解肉汤中培养了14小时。
- 根据制造商的说明,使用ZR BAC DNA Miniprep试剂盒(Zymo,目录号D4048)提取BAC DNA。
- 使用Qubit(ThermoFisher)对纯化的DNA进行定量。
- BAC DNA的处理类似于SHARE–seq ATAC–seq实验。
- 简而言之,将来自多个克隆的50纳克BAC DNA混合在一起,按照SHARE–seq转座条件进行处理。
- 经过处理的DNA使用Qiagen Minelute PCR清洁试剂盒进行纯化,然后进行七个循环的PCR扩增。
- 为了尽量减少批次效应,我们生成了五个生物学重复,并将所有材料汇集起来用于测序。
- 该文库在Nova-seq平台(Illumina)上使用100周期试剂盒进行测序(读取1,50周期;索引1,8周期;索引2,8周期;读取2,50周期)。
- 测序数据的处理方式与SHARE–seq ATAC–seq数据相同。
Generation of human genomic DNA data
人类基因组DNA数据的生成
Para_01
- 人类女性基因组DNA购自Promega(目录号G1521)。
- 基因组DNA用限制性酶SbfI-HF(新英格兰生物实验室,目录号R3642L)消化。
- 对于两个重复中的每一个,25μg的DNA在500μl反应体系中用200单位的SbfI-HF在37°C下过夜消化。
- 消化后的DNA在1%琼脂糖凝胶上电泳,切取对应2-2.5、2.5-3、3-4和4-5kb的片段。
- 所有来自重复1的片段以及来自重复2的3-4kb片段使用QIAquick凝胶提取试剂盒(Qiagen)纯化。
- 使用Zymo DNA清洁和浓缩试剂盒浓缩纯化产物。
- 最后,采用上述BAC DNA片段化处理方案进行标记和文库制备,在大小选择后回收到较少量的DNA时,使用相同的DNA质量与Tn5的比例。
In vitro footprinting
体外足迹分析
Para_01
- BAC DNA(50纳克)在室温下与重组c-MYC/MAX(Active Motif,目录号81087)或CEBPA(OriGene,目录号TP760685),标记缓冲液(20 mM Tris,10 mM MgCl2和20%二甲基甲酰胺)和水在总体积为40微升的反应体系中孵育1小时。
- 接下来,将由0.15微升预组装的Tn5(seqWell,Tagify(TM))、4.85微升稀释缓冲液(50 mM Tris,100 mM NaCl,0.1 mM EDTA,1 mM DTT,0.1% NP-40和50%甘油)和5微升标记缓冲液组成的混合物加入到样品中,在总体积为50微升的条件下于37°C孵育30分钟(最终TF浓度分别为0、50和100纳摩尔)。
- 通过Qiagen Minelute PCR纯化试剂盒纯化标记的DNA,并随后进行五轮PCR扩增。
- 然后将样品合并并在Nova-seq平台(Illumina)上测序。
- 测序数据处理方式与批量ATAC-seq数据相同。
Ageing multiome experiment
老龄化多组学实验
Para_01
- 鼠类实验得到了哈佛大学动物护理与使用委员会的批准,并严格遵守相关规定进行。
- C57BL/6小鼠从杰克逊实验室或国立老龄化研究所的老年啮齿动物群系(查尔斯河实验室)获得。
- 它们被安置在标准通风架上,每笼2到5只,提供自由饮食和饮水,在一个由实验室动物委员会认可的无特定病原体设施内饲养。
- 小鼠笼子里装有安德森的Bed-o'Cob垫料(安德森公司),两个窝垫(安卡公司,2×2英寸压缩棉方块)和一个红色小鼠屋(生物服务公司)。
- 为了HSC分离和流式细胞术,从年轻(n=10,11周大)和老年(n=5,24个月大)雄性C57BL/6小鼠的长骨(每只小鼠两根股骨和两根胫骨)的骨髓中用21号针头冲洗到染色介质(HBSS/2% FBS)中。
- 细胞沉淀后重新悬浮在ACK裂解缓冲液中,冰上放置5分钟。
- 小鼠数量根据预期的细胞产量和单细胞分析所需的输入量确定;来自小鼠的细胞混合在一起,因此没有进行盲法或随机化处理。
- 然后用染色介质洗涤细胞,通过40微米细胞过滤器,沉淀后在冰上与以下小鼠抗体制剂孵育30分钟:CD3克隆C145-2c11(宝利德,目录号100304;1:100),CD4克隆GK15(宝利德,目录号100404;1:400),CD5克隆53-7.3(eBioscience,目录号13-0051-85;1:400),CD8克隆53-6.7(宝利德,目录号100704;1:400),CD19克隆6D5(宝利德,目录号115504;1:400),B220克隆RA3-6B2(宝利德,目录号103204;1:200),GR1(Ly6-G/Ly6-C)克隆RB6-8C5(eBioscience,目录号13-5931-82;1:400),Mac1/CD11b克隆M1/70(宝利德,目录号101204;1:800)和Terr119克隆TERR-119(宝利德,目录号116204;1:100)。
- 然后在染色介质中洗涤细胞,为每个样品保留一小份作为未耗尽对照,并使用绵羊抗大鼠Dynabeads(赛默飞世尔科技,目录号1135)在磁铁上进行谱系耗尽。
- 细胞洗涤、沉淀后,在冰上与以下抗小鼠抗体混合物孵育45分钟以识别HSCs:太平洋橙链霉亲和素(赛默飞世尔科技,目录号S32365;1:500),PE/Cy7 Sca1(Ly-6a/E)克隆D7(eBioscience,目录号25-5981-82;1:200),APC cKit克隆2B8(BD Pharmingen,目录号553356;1:200),FITC CD48克隆HM48-1(宝利德,目录号103403;1:200)和PE CD150克隆Tc15-12F12.2(宝利德,目录号115904;1:200)。
- 孵育后,细胞洗涤并重新悬浮在染色介质中,并在流式细胞术前立即加入7-AAD(BD Pharmingen,目录号559925;1:50)。
- HSCs(活Lin− Sca1+ cKit+ CD48− CD150+)的细胞分选在BD FACS Aria II上进行。
- 同龄组内的细胞分选到同一管中,以便后续测序。
- 数据分析使用BD FACS Diva(v.8.0.2)和FlowJo(v.10.8.2)软件进行。
- 数据处理使用Cell Ranger ARC 2.0.0进行。
Para_02
- 核仁随后按照10x Genomics提供的低细胞输入核仁分离协议进行分离,该协议在CG000365用户指南中有描述。
- 然后使用10x Genomics的Chromium单细胞多组学ATAC+基因表达试剂盒(10x Genomics),根据制造商的说明处理核仁,每份样本获得2,000到10,000个细胞。
- 文库在Illumina Next-seq系统上进行测序,使用的测序格式如下:读取1,28;i7索引,10;i5索引,10;读取2,44(scRNA-seq);读取1,30;i7索引,8;i5索引,24;读取2,30(scATAC-seq)。
- 数据处理使用了10x Genomics的CellRanger ARC 2.0.0软件。
Tn5 sequence bias modelling
Tn5 序列偏差建模
Getting Tn5 insertion counts
获取Tn5插入计数
Para_01
- 片段文件的末端通过+4/-4进行移动,以获得Tn5转座所形成的9 bp交错末端的中心。然后对每个样本的每个cCRE中的每个单碱基位置的插入次数进行量化,并存储在一个样本- cCRE-位置三维张量中,以便快速检索数据。
- 这样可以迅速获取数据。
Data preprocessing
数据预处理
Para_01
- 该模型将局部DNA序列环境作为输入,并预测单碱基分辨率的Tn5偏差。
- 为此,每个感兴趣位置周围的±50-bp DNA序列通过一对一编码转换为101×4矩阵,并用作模型输入。
- 对于预测目标,我们使用局部相对Tn5偏差作为目标值。
- 更具体地说,每个位置的原始Tn5插入计数除以±50-bp窗口内的平均Tn5插入计数。
- 低局部覆盖度(每碱基对少于20次插入)的位置被移除,以确保训练数据的质量。
- 为了便于模型训练,将得到的观测Tn5偏差值进行log10变换并重新缩放。
- 在数据集划分方面,我们将所有BAC随机划分为80%、10%和10%,分别用于训练、验证和测试集;换句话说,来自同一BAC的所有数据属于同一分区。
- 这样做是为了防止重叠的局部序列环境同时出现在训练和测试数据集中,这可能导致性能高估。
- 为了保证不同偏差水平的例子具有相同的覆盖率,我们将所有训练例子基于其Tn5偏差值分为五个区间,并对每个区间进行上采样,使得所有区间最终具有相同数量的例子。
- 此外,考虑到Tn5插入的对称性,我们将训练例子的反向互补序列作为数据增强。
- 原始数据和反向互补数据组合后,进行了打乱,然后用于模型训练。
Model architecture
模型架构
Para_01
- 卷积网络由三个卷积和最大池化层以及两个全连接层组成。
- 每个卷积和最大池化层依次执行卷积、修正线性单元(ReLU)非线性激活和最大池化。
- 我们为每层使用了宽度为5的32个滤波器,并且使用了‘same’填充模式和步长大小为1。
- 随后的两个全连接层的输出维度分别为32和1;第一个全连接层使用ReLU激活函数,第二个(即最终输出层)使用线性激活函数。
Model training and evaluation
模型训练和评估
Para_01
- 该模型是在训练集上进行训练的,并且超参数是基于验证集上的性能进行优化的。冻结模型的最终性能是在测试集上进行评估的。
- 该模型是使用Keras61实现的,以均方误差作为损失函数,并使用带有默认参数的Adam优化器62进行优化。
- 训练是以批量大小为64和基于验证集上的模型损失进行提前停止的方式进行的。
Benchmarking with other Tn5 bias models
与其他Tn5偏差模型的基准测试
Para_01
- 包括 k-mer 模型(k = 3, 5, 7)和 PWM 方法(单核苷酸和二核苷酸)在内的方法被纳入基准测试。
- 对于 k-mer 方法,量化了所有可能的 k-mer 序列的前景频率和背景频率。
- 前景/背景频率比被用作相应 k-mer 的 Tn5 偏好估计。
- 对于单核苷酸 PWM,我们在±10-bp 窗口内(总长度为 21 bp)计算前景和背景碱基频率,并计算 Tn5 插入的 PWM。
- 使用 TOBIAS18(默认设置)计算二核苷酸 PWM 分数。
- 建议为每个数据集训练自定义的 ChromBPNet 偏差模型,以代表 Tn5 序列偏好并实现最高质量的模型。
- 因此,我们使用 HepG2 和 K562 的 ATAC-seq 数据训练了 ChromBPNet 来评估其推荐设置下的性能。
Genome-wide Tn5 bias reference tracks
全基因组Tn5偏差参考轨迹
Para_01
- 人类基因组(hg38)、小家鼠基因组(mm10)、果蝇基因组(dm6)、酿酒酵母基因组(sacCer3)、秀丽隐杆线虫基因组(ce11)、斑马鱼基因组(danRer11)和黑猩猩基因组(panTro6)的序列从UCSC基因组浏览器网站(https://hgdownload.soe.ucsc.edu/goldenPath/)下载。
- 上述Tn5偏差神经网络模型被应用于参考基因组中的每个位置,以生成全基因组范围内的Tn5偏差轨迹。
Computation of footprint scores
足迹分数的计算
Para_01
- 为了检测染色质开放区(cCREs)中不同尺度下的DNA-蛋白质相互作用,我们实现了一个框架来计算每个碱基对位置的足迹评分。
- 简而言之,对于每个单独的碱基对位置,我们定义一个中心足迹窗口以及两侧的翼侧窗口(图1a)。
- 然后我们计算了中心/(中心+翼侧)Tn5插入计数的观测比率。
- 将前景的观测比率与背景分布进行比较以计算统计显著性,然后将其转换为足迹评分。
Estimation of background dispersion
背景扩散的估计
Para_01
- 给定特定的中心偏差、侧翼偏差和局部覆盖组合,我们预计在没有蛋白质结合的情况下,中心/(中心+侧翼)插入比例将呈现某种分布;这被定义为背景分布。
- 这种背景分布可以通过BAC裸DNA Tn5插入数据来估算。
- 为此,我们首先从BAC数据集中随机采样了100,000个位置,并检索了它们的局部覆盖度(定义为中心和侧翼区域的总插入数)、中心偏差和侧翼偏差。
- 然后,对于每个采样的位置A,我们在三维空间中的中心偏差、侧翼偏差和局部覆盖度中识别出最近邻的500个位置(NN1–NN500)。
- 为了确保每个维度都被等权重处理,首先将每个维度的值标准化为零均值和单位方差。
- 这500个最近邻观测可以被视为具有几乎相同偏差和覆盖度的背景观测,NN1–NN500的中心/(中心+侧翼)比率构成了位置A的背景分布。
- 因此,对于这100,000个采样的每个位置,我们可以计算其背景比率分布的平均值和标准差。
- 这使我们能够训练一个背景离散模型,该模型以(中心偏差、侧翼偏差和局部覆盖度)作为输入,并有效地预测背景分布的平均值和标准差。
- 为了确保模型接触到广泛的局部覆盖度的训练样本,我们将BAC数据集下采样至原始测序深度的50%、20%、10%、5%和1%。
- 最后,我们训练了一个具有单个隐藏层(32个节点,ReLU激活函数)和线性输出层激活的神经网络。
- 数据集被随机拆分为80%用于训练,10%用于验证和10%用于测试。
- 该模型使用Keras实现,并使用均方误差损失函数通过Adam优化器在训练数据集上进行训练。
- 通过验证集上的损失确定提前停止,最终模型的性能在测试集上进行了评估。
- 此外,由于足迹半径变化时总中心或侧翼偏差存在显著差异(补充说明),我们为每个足迹半径分别训练了单独的模型。
Calculation of footprint scores
足迹分数的计算
Para_01
- 对于 cCRE 中的每个位置,我们定义一个中心足迹窗口和两侧的侧翼窗口。
- 我们首先计算 Tn5 插入计数的前景观察到的中心/(中心+侧翼)比率,然后应用预训练的背景分散模型来计算其背景分布的均值和标准差。
- 接下来,我们使用单尾 z 检验来计算足迹的 P 值。
- 如果观察到的比率明显低于背景分布,则该位置可能被蛋白质结合。
- 更具体地说,为了避免在仅有一侧侧翼显示比中心窗口更高的 Tn5 插入的位置上标记足迹,我们分别进行中心对左侧和中心对右侧的测试,并保留较高的 P 值(补充说明)。
- −log10(P 值) 通过运行最大值和平滑平均值平滑处理,然后用作最终的足迹评分。
Aggregate footprinting
聚合足迹
Para_01
- 为了计算总足迹,首先对基因组中围绕转录因子或核小体结合位点的Tn5插入进行聚合,然后用于计算足迹评分。
- 对于转录因子,我们选择了具有匹配转录因子基序的位点,使用motifmatchr版本1.24.0(参考文献65)(P截止值1×10^-5),以及与相应转录因子的ChIP-seq峰重叠的位点。
- 对于反向链上的基序匹配,将围绕该基序的Tn5插入图谱反转,以便不同位点的插入在相同方向上对齐。
Footprint-to-TF prediction
足迹到张量预测
Para_01
- seq2PRINT被选为我们主要的TF结合预测器,但我们仍然提供了这种轻量级的足迹到TF预测模型以发挥其速度优势。
- 有关足迹到TF预测与基于seq2PRINT的TF结合预测之间的比较,请参阅补充说明中的'多尺度足迹预测TF的多种方法'。
Para_02
- 为了预测转录因子(TF)结合的景观,我们训练了一个二元分类器来预测任何TF基序位点是否被相应的TF结合。
- 基序位点通过motifmatchr软件包中的matchMotifs函数识别。
- 所有匹配P值低于5×10^-5的位点都被保留。
- 对于任何TF基序位点,我们使用多尺度(20、40、60、100、160或200-bp直径)足迹,在围绕基序中心的±100-bp局部区域内进行分析,以及作为模型输入的基序匹配得分。
- 由matchMotifs函数返回的基序匹配得分被转换为均匀分布。
- 因此,通过将来自六个不同尺度的201维足迹向量与单个基序匹配得分相结合,我们创建了一个1,207维的向量作为最终模型输入。
- 足迹分数的前1,206维被单独标准化为零均值和单位方差。
- 对于预测目标,我们将与同一TF的ChIP峰重叠的所有位点标记为1,而那些不与ChIP重叠的位点标记为0。
- 发现一些TF的基序位点与ChIP重叠的比例非常低(10%或更低),这可能是由于基序或ChIP数据集的质量较低,这些TF被从模型训练和测试中移除。
- 我们还添加了额外的负样本以及反向互补样本以进行数据增强。
Para_03
- HepG2数据被用作训练数据,GM12878数据(先前在原始SHARE–seq论文27中发表)作为验证。
- 在固定模型超参数后,HepG2和GM12878数据被合并成一个更大的训练数据集,以训练最终的足迹到TF模型。
- 最终模型在K562 scATAC数据66以及人类BMMC SHARE–seq数据集中的三种细胞类型( naïve B细胞、CD14单核细胞和晚期红细胞系细胞)上进行了测试。
Model architecture and training
模型架构和训练
Para_01
- TF结合预测模型是一个具有两个隐藏层(128+32个节点)的神经网络模型。
- 两个隐藏层均使用ReLU激活函数,最终输出层使用sigmoid激活函数。
- 该模型使用Keras实现,并通过Adam优化器使用大小为128的批次在训练数据集上进行训练。
- 二元交叉熵被用作损失函数。
- 基于验证集上的模型损失,使用了提前停止策略。
ChIP validation and benchmarking with previous methods
使用先前的方法对ChIP进行验证和基准测试
Para_01
- 为了评估模型性能,我们使用了ChIP–seq作为真实情况,并验证了预测的结合事件。
- HepG2和GM12878数据从UniBind68(https://unibind.uio.no/search)下载用于模型训练。
- BMMC细胞类型的ChIP–seq数据从cistromeDB69下载。
- 为了与先前的方法进行基准测试,为了确保我们只包括高质量的TF结合位点,我们从UniBind下载了K562基于ChIP的TF结合数据。
- 对于cistromeDB数据集,我们应用了网站上指定的质量控制过滤器(http://cistrome.org/db/#/about)。
- 更具体地说,我们应用了以下过滤器:FRiP 0.01或更高,FastQC 0.25或更高,唯一映射比例0.6或更高,折叠变化超过十倍的峰,峰数超过500,峰与DNase I超敏感位点比率0.7或更高,以及PCR瓶颈系数0.8或更高。
- 包括以下细胞类型标签的数据集:单核细胞、B淋巴细胞、红细胞、红细胞祖细胞和红细胞前体。
- 如果同一TF存在多个数据集,则保留所有相同TF数据集的交集作为最终的高置信度结合位点列表,用于模型训练。
Para_02
- UniBind中的K562数据集被用于与先前的方法进行基准测试,包括从ChromBPNet获得的DNase I足迹分析、TOBIAS和序列归因得分。
- 简而言之,所有ATAC足迹分析方法和ChromBPNet都使用相同的ATAC–seq数据作为输入。
- 而对于K562的DNase I足迹分析结果,则是从ENCODE数据集ENCLB253REF、ENCLB843GMH和ENCLB096YUZ中获得(参考文献11)。
- 为了保证公平比较,我们使用了同一组基序匹配位置(先前发表于参考文献70)作为候选结合位点,并将每种方法预测的分数映射到这些位点上进行比较。
- 对于具有多个UniBind数据集的转录因子,它们的交集被用于基准测试。
- 然后,对于每种方法,我们根据预测的结合分数对候选位点进行排名,并使用前10%的位点来评估预测的准确性。
- 通过Gviz包(v.1.46.1)71可视化预测的和真实结合位点。
- 此外,为了评估每个模型的假阳性率,我们还在我们的BAC裸DNA数据上测试了所有三种基于ATAC的模型。
- 每个模型都使用相同的数据作为输入,预测的结合事件数量用来表示假阳性预测。
Footprint-to-nucleosome prediction
足迹到核小体预测
Input data
Para_01
- 为了预测核小体占用情况,我们训练了一个回归神经网络模型。
- 对于任何基因组位置,我们将直径为20、40、60、100和160 bp的多尺度足迹作为输入提供给该模型,范围在±100 bp的局部区域内。
- 未包括200 bp尺度,以防止模型学习相邻核小体的同时占据。
- 为了训练此模型,我们使用了之前发表的化学映射的酿酒酵母核小体占用数据作为训练标签。
- 并使用之前发表的酿酒酵母ATAC-seq数据计算多尺度足迹作为训练输入。
- 我们在局部ATAC-seq覆盖度超过10的区域保留观察数据,并将核小体占用值的第5和第95百分位分别重缩放为0和1。
- 在数据分割方面,我们随机按染色体将所有数据划分为训练集(chrVII, chrXI, chrIX, chrI, chrV, chrX, chrVIII 和 chrXII),验证集(chrIV 和 chrII)和测试集(chrVI, chrXVI, chrXIII, chrIII, chrXIV 和 chrXV)。
- ,
Model architecture and training
模型架构和训练
Para_01
- 核小体预测模型是一个具有两个隐藏层(64 + 16个节点)的神经网络模型。
- 两个隐藏层均使用ReLU激活函数,最终输出层使用线性激活函数。
- 该模型使用Keras实现。
- 该模型使用Adam优化器,在训练数据集上以批量大小为128进行训练。
- 均方误差作为损失函数。
- 根据验证集上的模型损失,使用了提前停止策略。
Performance evaluation
绩效评估
Para_01
- 模型性能通过上述测试酵母染色体上的数据进行评估。
- 总共使用了测试酵母染色体(chrVI、chrXVI、chrXIII、chrIII、chrXIV 和 chrXV)上长度为 5 kb 的 859 个区域进行测试。
- 我们检测了预测的核小体信号峰和真实核小体占用情况,分别作为预测和观察到的核小体。
- 精确度被计算为具有在一定截断距离内(本研究中为 50 或 75 bp)的真实核小体的预测核小体所占的百分比。
- 召回率被计算为具有在相同截断距离内预测核小体的真实核小体所占的百分比。
Seq2PRINT
Seq2PRINT
Model architecture and training
模型架构和训练
Para_01
- seq2PRINT 模型是一个卷积神经网络,它将 one-hot 编码的 DNA 序列(长度为 L 的 DNA 序列编码成一个 L × 4 的矩阵,其中每一行有一个元素设置为 1,表示特定的核苷酸)作为输入,并预测相应的多尺度足迹,在碱基对分辨率上。
- 该架构如扩展数据图 4a 所示。
- 为了便于阐述架构,我们将 seq2PRINT 模型分为三个部分。
- 尽管 seq2PRINT 的架构可以根据可用的计算资源以及训练数据的深度和规模灵活调整,但在本文的所有结果中,我们构建了模型并选择了以下超参数。
Para_02
- 第一卷积层由1,024个宽度为21 bp的滤波器组成,并使用高斯误差线性单元(GELU)激活函数,旨在从输入的DNA序列中捕捉信息性的序列模式(即序列基序)。
- 随后,输出被传递到八个具有残差连接的卷积块层。
- 每个卷积块由一个分组扩张卷积层组成(n个滤波器=1,024,宽度=3,组数=8,扩张率=2i,i=1,…,8),接着是一个逐位置前馈层(实现为一个宽度为1的卷积层,n个滤波器=1,024)。
- 在这些卷积块之间插入了带有GELU激活函数的批量归一化层。
- 扩张率的增加导致神经网络的接受域扩大,从而捕捉序列模式与其上下文之间的关系。
- 通过八个卷积块层,seq2PRINT对每个cCRE的接受域为±920 bp。
- 使用分组卷积层可以构建更宽的模型,以有限的参数捕获更多的信息,提供了正则化效果和减少计算复杂度。
- 最后,卷积块堆叠的输出被传递到输出层。
Para_03
- 在这篇论文中,我们设计了两个输出层,一个多尺度足迹层(滤波器宽度为1的卷积层),用于输出多尺度足迹,以及一个可及性层(全局平均池化层后接全连接层),用于预测特定cCRE中的Tn5插入数量。
Para_04
- 为了便于seq2PRINT模型的训练,我们在图形处理单元上实现了批处理高效的多尺度足迹计算,该计算遵循与所述足迹计算相同的数学模型,唯一的区别在于它输出z统计量而不是从z检验计算出的P值。
Para_05
- 在训练过程中,模型权重被更新以最小化以下损失函数:
Para_06
- 其中MSE表示均方误差,MSE(x,y)=∑(x-y)²,footprint_PRINT和footprint_pred分别代表由PRINT框架和seq2PRINT模型计算出的多尺度足迹,nobs和npred分别代表该区域内的观测和预测的Tn5插入。
- 其中PRINT表示多尺度足迹,footprint_PRINT和footprint_pred分别代表由PRINT框架和seq2PRINT模型计算出的多尺度足迹,nobs和npred分别代表该区域内的观测和预测的Tn5插入。
Para_07
- 值得注意的是,在卷积块之前,访问层的梯度反向传播被中断了:换句话说,seq2PRINT模型前面几层学习到的序列模式和它们之间的关系完全由多尺度足迹目标驱动。
- 访问输出层和相应的损失函数仅用于解释这些已学习的序列特征(有关详细说明,请参阅以下部分)。
- 这种设计消除了在足迹损失和访问损失之间选择权重的需要,并使seq2PRINT成为一种由足迹驱动的序列模型,从而将其与以前的由访问驱动的模型区分开来(例如,Basset73)。
Para_08
- seq2PRINT 模型使用了学习率为 3 × 10−4 的 Adam 优化器,并采用指数移动平均来稳定和改进模型收敛。
- 在这项研究中,我们采用了基于染色体的五折交叉验证,各折的输出结果取平均作为最终预测。
Deriving sequence attribution scores
推导序列归属分数
Para_01
- 我们使用DeepLIFT74方法计算序列归因分数,这代表了给定输入DNA序列中的每个碱基对如何对从训练过的seq2PRINT模型得到的具体标量输出做出贡献。
- 可访问性层的输出是每个区域的一个标量,这使其自然适合成为DeepLIFT计算归因分数的目标。
- 然而,多尺度足迹层不是一个标量而是一个大小为L×尺度数量的矩阵。
- 因此,我们设计了两种策略将输出足迹总结为一个标量值。
Para_02
- 这两种策略都涉及将预测的z统计量转换为对数(P值)足迹得分,如同PRINT框架所示。
- 第一种策略涉及手动检查,如图2b所示,在观察到和预测的多尺度足迹中定位感兴趣的区域×尺度,计算该区域内足迹得分之和并计算相应的序列归因得分。
- 第二种策略是在整个峰区域内求和足迹得分,更适合全基因组计算。
- 如果没有进一步说明,我们所说的从可及性层计算出的序列归因得分为计数序列归因得分,而从足迹层计算出的序列为足迹序列归因得分。
Para_03
- 对于本文档中的所有结果,我们使用20个二核苷酸洗牌的输入序列为DeepLIFT算法的参考序列。
- 我们将DeepSHAP包中的DeepLIFT算法实现进行了改编,以适应此框架中使用的自定义非线性函数;自定义实现作为https://github.com/buenrostrolab/scPrinter的一部分可用。
De novo motif identification based on sequence attribution scores
基于序列归因分数的新motif识别
Para_01
- TF-MoDISco(tfmodisco-lite v.2.2.1)被用来基于序列归因得分推断新的基序。
- 简而言之,TF-MoDISco识别输入序列中具有高序列归因得分(seqlets)的局部区域,然后将相似的seqlets对齐和聚类成组的新基序。
- 我们将seqlets的数量设置为1,000,000,并保持其余参数为默认值。
- 通过TomTom(meme套件v.5.5.7)将新发现的基序分配给已知基序。
- 为了推断新基序与cCREs的匹配情况,我们使用软件finemo(提交编号830d7f3),该软件接受新基序和序列归因得分。
TF binding prediction using attribution scores
使用归因得分预测TF结合
Para_01
- 计算得到的序列归属分数突出了影响足迹和可及性的转录因子结合位点。
- 因此,我们训练了一个二元分类器,它类似于足迹到转录因子模型,但与多尺度足迹不同,它接受计数和足迹序列归属分数。
Para_02
- 训练和验证的TF结合位点与之前的足迹到TF模型保持一致。对于每个基序位点,特征包括围绕该基序中心±100 bp区域内的计数和足迹序列归属评分、匹配基序的得分以及关注基序与匹配位点处序列归属评分之间的皮尔逊相关性,形成一个405维的向量。
Para_03
- TF绑定模型包含三个隐藏层(256、128和64个神经元),中间使用了GELU激活函数和0.25的dropout比率。
- 该模型使用Adam优化器进行训练,目标函数为二元交叉熵。
Para_04
- 这个TF结合模型是我们用于图2至图4的最终TF结合预测模型,因为它具有优越的性能。
- 关于足迹到TF预测与基于seq2PRINT的TF结合预测之间的比较,请参阅补充说明中的‘多尺度足迹预测TF结合的多种方法’。
LoRA enables efficient sequence modelling for scATAC–seq data
LoRA能够对scATAC-seq数据进行高效的序列建模
Para_01
- 为了使序列模型在具有多种细胞类型或细胞状态的scATAC-seq上更具可扩展性,我们使用了LoRA技术对神经网络模型进行高效的参数微调。
- 具体来说,对于给定的scATAC-seq数据,我们首先通过聚合数据集中所有细胞的Tn5插入来训练一个seq2PRINT模型(称为预训练模型)。
- 接下来,对于每个根据特定细胞状态聚集的伪批量细胞,我们使用LoRA微调技术推导出一个特定于该伪批量的seq2PRINT模型。
- 我们描述了这个微调过程如下。
Para_02
- 对于任何由初始权重 W0 参数化的神经网络层,其中 (W_{0} \in R^{d_{p"}}),LoRA 模型将学习一个更新参数 (\Delta W \in R^{d_{p"}}),其中 dp 是该层的可学习参数的数量,这两个参数的和被用作结果微调神经网络的权重参数 (W = W_{0} + \Delta W)。
- 对于我们用于 seq2PRINT 的残差分组卷积层,
Para_03
- 如果我们分别针对 n 个感兴趣的伪批量微调模型,这将导致总共需要学习 n × dp 个参数。
- let's think step by step.
错误!!! - 待补充
Predicted impact of TF motifs on footprints
TF基序对足迹预测影响
Para_01
- 为了探讨seq2PRINT模型学习到的多尺度足迹与DNA序列之间的关系,我们从seq2PRINT生成了给定已知或新发现基序的边际预测。
- 对于一个新发现的兴趣基序,我们首先通过在每个位置取最高概率的核苷酸来确定其共识序列。
- 然后,我们从数据集中随机选择了25,000个cCRE,并将共识序列置于中心,在采样的cCRE周围平均模型预测。
- 对于已知的基序,我们收集其在cCRE中的匹配位置,打乱这些位置并平均模型预测。
- 在这两种方法中,我们都将给定基序的存在与否之间的差异作为边际预测进行计算。
Tracking TF binding dynamics across human haematopoiesis
追踪人类造血过程中TF结合的动力学
Generation of pseudo-bulks
伪批量的生成
Para_01
- 人类BMMC数据集中的单细胞首先使用cisTopic57嵌入到较低维度的空间中,然后根据它们在cisTopic空间中的空间邻近性被分组成1,000个伪批量。
- 更具体地说,我们首先采样了1,000个细胞作为伪批量中心,然后在cisTopic空间中识别每个中心细胞的k-最近邻(KNN,k = 5,000)作为同一伪批量的其他成员。
- 我们认为,通过防止对密集连接的局部邻域进行过度采样,用低局部连通性的中心细胞进行采样可以帮助增加状态空间的覆盖范围。
- 因此,我们首先随机采样了10,000个支架细胞,并使用这些细胞构建了一个KNN图(k = 10),然后选择了KNN图中最低入度的1,000个细胞作为伪批量中心。
Computing pseudo-time
计算伪时间
Para_01
- 伪时间沿着人类造血谱系计算使用了Palantir软件包(版本1.0.0)。
- 为了减少计算时间,我们从人类BMMC数据集中随机抽取了100,000个细胞作为支架细胞。
- 支架细胞和伪批量中心细胞的cisTopic嵌入被用作Palantir的输入。
Footprinting repressor TFs
足迹抑制因子TFs
Para_01
- 因为BHLHE40在T细胞中的作用已经得到了广泛的研究,所以扩展数据图2中的BHLHE40足迹分析结果是使用来自人类骨髓数据集的CD8+ T细胞获得的。
TF binding in erythroid and lymphoid cell cCREs
TF在红系和淋巴系细胞cCRE中的结合
Para_01
- 对于红系和淋巴系发育轨迹,我们首先通过选择RNA水平与伪时间的相关性大于0.5的基因来识别相关基因。
- 然后,我们在相关基因的转录起始位点±50 kb范围内筛选cCREs,并保留那些cCRE可及性和伪时间之间的相关性大于0.5的cCREs。
- 对于所有cCREs,我们通过保留那些seq2PRINT TF结合评分与伪时间之间的相关性高于0的TF结合位点来定位发育过程中激活的候选TF结合位点。
- 剩余的TF结合位点用于动态量化。
- 为了量化推断的结合时机,我们将每个结合位点的TF结合评分重新缩放到[0,1]范围,并计算曲线下面积,较高的值表示结合信号上升得更早,反之亦然。
PCA measures TF binding pattern complexity at each cCRE
PCA在每个cCRE处测量TF结合模式的复杂性
Para_01
- 为了揭示人类BMMC数据集中每个cCRE在不同细胞群体中的TF结合模式的复杂性,我们使用了一种基于主成分分析(PCA)的方法。
- 为了减少计算复杂度,我们首先将1,000个经过LoRA微调的seq2PRINT模型(对应1,000个伪整体样本)压缩成20个模型,分别对应20种细胞类型。
- 在这个过程中,对应相同细胞类型的伪整体样本的模型权重被平均。
- 然后,我们为这20种细胞类型生成了整个cCRE的TF结合评分。
- 我们将每个cCRE用10-bp窗口进行分割,并在每个窗口内平均TF结合评分。
- 随后,对每个cCRE的这个20 × 10-bp窗口矩阵使用PCA作为降维方法,使用解释98%方差的最少数量的主成分来量化TF结合模式的复杂性。
Characterizing age-related cCRE reorganizations
描述与年龄相关的cCRE重组
Data preprocessing
数据预处理
Para_01
- 细胞的FRIP低于0.3且深度小于300首先被移除。
- 此外,我们使用ArchR(https://www.archrproject.com/)79计算每个单细胞的双重分数,并移除了前5%的双重分数较高的细胞。
- 剩余的细胞随后使用Seurat V3包(v.5.0.3)80进行处理。
- 细胞通过潜在语义索引嵌入到低维空间中,然后进行聚类。
- 选择与HSC相对应的Seurat聚类进行伪集合并进行下游差异测试。
- 具有LinNeg FACS分选标签的细胞被排除在HSC特异性分析之外。
- 为了识别代表性细胞状态,我们使用SEACells(https://github.com/dpeerlab/SEACells/tree/main)82识别了HSC中的100种代表性细胞状态。
- 代表性细胞用作中心形成伪集簇。
- 每个伪集簇通过按距离递增顺序从中心细胞包含最近邻细胞,直到总共有500万个片段为止来生成。
Defining HSC subpopulations
定义HSC亚群
Para_01
- 从10x Multiome获得的单细胞RNA表达数据首先通过去除前5%高表达基因以及核糖体和线粒体基因进行了过滤;移除了少于100个RNA计数的细胞。
- 然后我们使用SCTransform(v.0.4.1)运行了5,000个可变特征来对数据进行归一化。
- 归一化后的值被用作输入传递给Spectra83(https://github.com/dpeerlab/spectra)。
- 接下来我们运行了Spectra,初始化时使用了默认的HSC和全局通路,并包括了从已发表文献中获取的额外基因集;去除了包含少于五个基因的基因集。
- 我们计算了每个Spectra程序在每个伪批量中的表达。
- 为此,我们使用DESeq2对伪批量按基因的RNA计数矩阵进行归一化,然后重新缩放每个基因的值。
- 对于任何特定的Spectra程序,我们生成了100个背景程序,这些程序由具有匹配总体表达水平的基因组成。
- 然后将感兴趣的Spectra程序的平均基因表达与采样的背景程序中的基因表达进行比较,以得出z分数。
- 最后,使用伪批量按程序的z分数矩阵将伪批量聚类为HSC亚群。
Differential testing
差分测试
Para_01
- 使用 DESeq2(版本 1.42.1)84 进行了差异 RNA 和 cCRE 可访问性测试。对于每个伪批量样本,我们量化了每个基因的总 RNA 读取计数以及每个峰内的 Tn5 插入计数(调整大小为 1 kb),并使用 DESeq2 识别显著的差异基因和 cCRE,年龄作为协变量。
- 对于每个伪批量样本,我们量化了每个基因的总 RNA 读取计数以及每个峰内的 Tn5 插入计数(调整大小为 1 kb),并使用 DESeq2 识别显著的差异基因和 cCRE,年龄作为协变量。
Predicting Ets/Runx dimer structures using Alphafold3
使用 Alphafold3 预测 Ets/Runx 二聚体结构
Para_01
- 我们为每个新识别的代表Ets/Runx复合基序的基序生成了共识DNA序列。
- 然后,我们将与PDB-4L0Z中相同的氨基酸序列段(Runx,54–212;Ets,332–432)以及共识DNA序列及其反向互补序列输入到Alphafold3中。
- 对于验证的二聚体结构对应的结构,我们使用PyMol(v.2.6)将AF3预测的结构与PDB结构进行比对,并计算均方根偏差。
- 所有结构均使用ChimeraX v.1.8进行可视化。
- 使用ChimeraX界面功能和默认参数识别溶剂不可及碱基。
Data availability
Para_01
- 原始和处理后的测序数据在这个研究中可以在基因表达全景图(GEO)上获取,访问编号为GSE216464。
- 预训练的机器学习模型和预先计算的Tn5偏差轨迹等附加资源可以在Zenodo上获得(https://doi.org/10.5281/zenodo.7121026)。
- 人类骨髓和小鼠老化数据集的交互可视化以及seq2PRINT足迹和转录因子预测的轨迹可以通过https://github.com/buenrostrolab/PRINT获取。
- H. sapiens(hg38)、M. musculus(mm10)、D. melanogaster(dm6)、S. cerevisiae(sacCer3)、C. elegans(ce11)、D. rerio(danRer11)和P. troglodytes(panTro6)的参考基因组可以在https://hgdownload.soe.ucsc.edu/goldenPath/获取。
- 来自GM12878的SHARE-seq数据可以从GEO(GSE140203)获取。
- 核小体的化学映射可以从GSE97290获取。
- ChIP-exo数据来源于GSE151287。
- 转录因子ChIP-seq数据可以从UniBind(https://unibind.uio.no/static/data/20220914/bulk_Robust/Homo_sapiens/damo_hg38_all_TFBS.tar.gz)和ENCODE(补充表8中列出了完整的访问编号列表)获取。
- CTCF降解数据可以从ENCODE(ENCSR328JGW, ENCSR260SWI)获取。
- 地塞米松处理的数据可以从ENCODE(补充表8中列出了完整的访问编号列表)和EMBL BioStudies(E-MTAB-9910, E-MTAB-9911 和 E-MTAB-9912)获取。
- 干扰素处理的数据可以从GEO(GSE75306)获取。
- 本研究中使用的所有资源可以在补充表8中找到。
Code availability
Para_01
- PRINT 和 seq2PRINT 的 Python 实现代码可以在 https://github.com/buenrostrolab/scPrinter 访问。
- 用于重现分析的代码和笔记本可以在 https://github.com/buenrostrolab/PRINT 找到。
- 文章中特定分析所用代码的静态版本可以在 Figshare (https://figshare.com/s/e173a06b0c62c2f9dd1c) 获得。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-02-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号