GRSA富集:可视化天花板你值得拥有


常用的功能富集分析方法,根据其所采用的统计方法大致可分为三类:
- (i)过度表达分析(ORA)
- (ii)功能类别评分(FCS)
- (iii)基于通路拓扑结构(PT)
最近我们介绍了好几种功能富集分析的包,这次又来了一个,个人比较喜欢的,里面可以绘制各种高分文章中出现的富集结果精美展示图,来看看~
文献信息:
Chen Peng, Qiong Chen, Shangjin Tan, Xiaotao Shen, Chao Jiang, Generalized reporter score-based enrichment analysis for omics data, Briefings in Bioinformatics, Volume 25, Issue 3, May 2024, bbae116, https://doi.org/10.1093/bib/bbae116
ReporterScore工具地址:https://cran.r-project.org/web/packages/ReporterScore
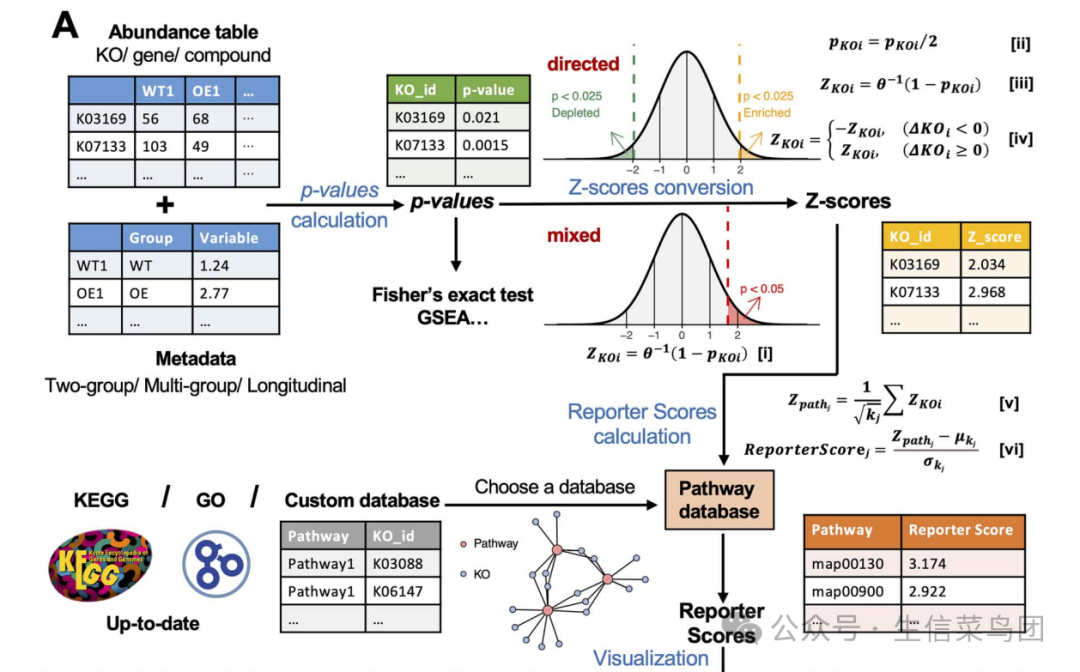
GRSA算法
(1) Calculating the P-values 计算pvalue
pvalue计算方法有以下这些:t检验,wilcoxon秩和检验,ANOVA检验等,更多见supplementary Table S1
(2) pvalue转换为z-score
(3) 对通路进行打分
(4) 最后进行可视化
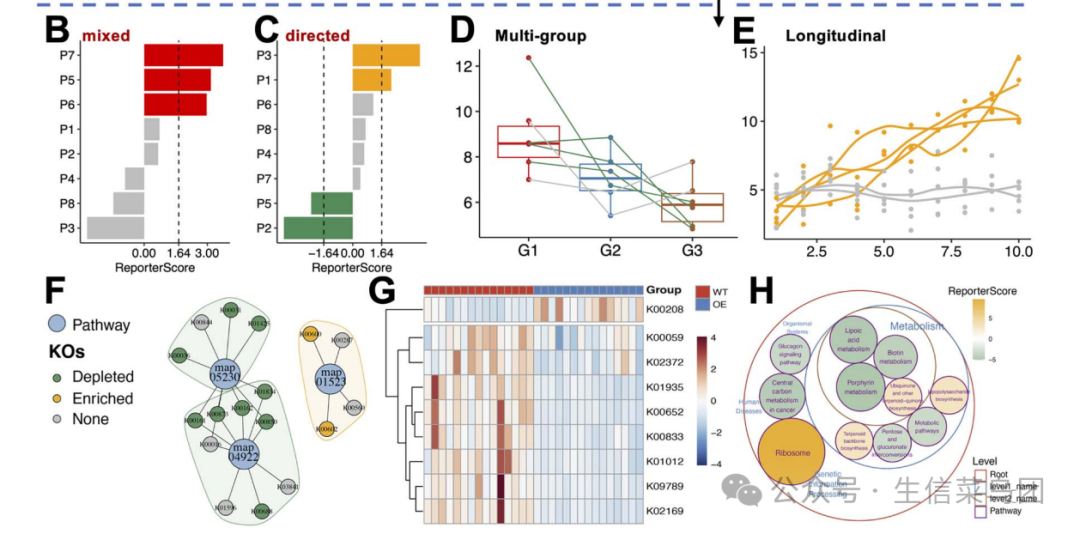
GSRA算法性能比较
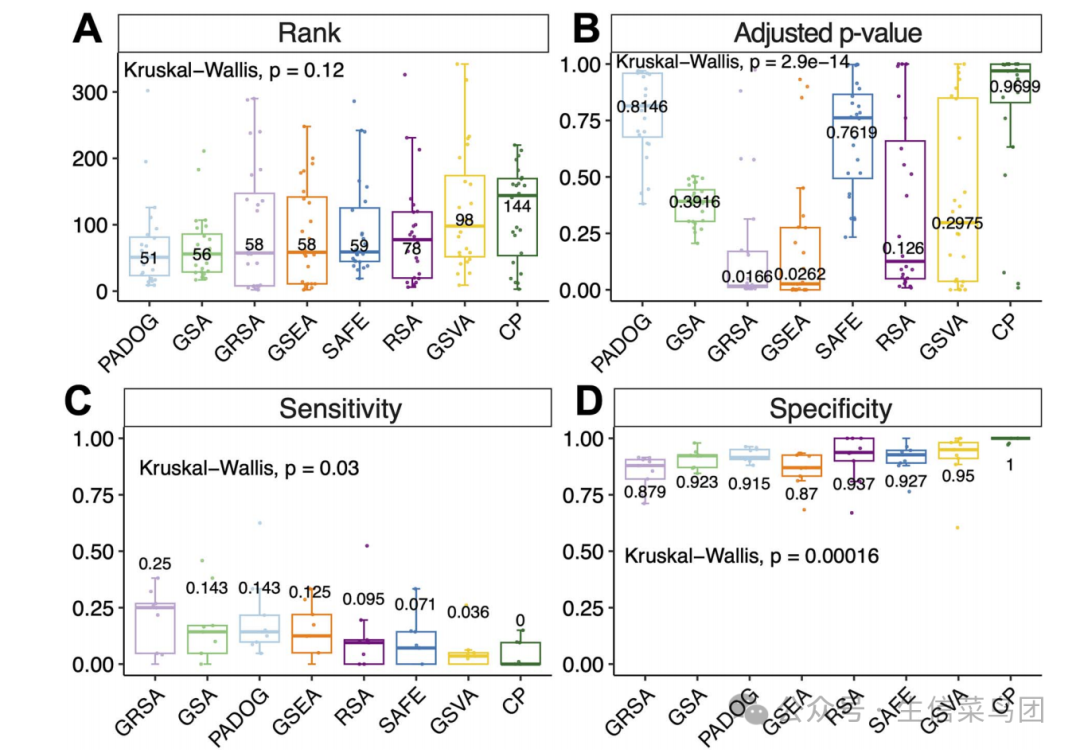
使用相同的通路数据库(KEGG v109.0)评估GRSA与其他流行的富集分析方法的性能:GRSA在无阈值的FCS方法中表现相当好,优于传统的ORA方法。
案例使用
1、软件安装:
# 安装cran上的包
options(BioC_mirror="https://mirrors.westlake.edu.cn/bioconductor")
options("repos"=c(CRAN="https://mirrors.westlake.edu.cn/CRAN/"))
install.packages("ReporterScore")
# 或者github上
# install.packages("devtools")
devtools::install_github("Asa12138/pcutils")
devtools::install_github("Asa12138/ReporterScore")
# 检测安装成功与否并加载
library(ReporterScore)
2、输入数据要求
数据为feature丰度数据如基因表达矩阵和样本metadata信息
- 对于转录组学、单细胞RNA测序(scRNA-seq)以及相关的基于基因的组学数据:可以使用完整的基因丰度表。
- 对于宏基因组学和宏转录组学数据:可以使用KO(KEGG Orthology)丰度表,该表通过使用Blast、Diamond或KEGG官方映射软件将读取序列或contigs与KEGG或EggNOG数据库进行blast来生成。
- 对于代谢组学数据:可以使用注释过的化合物丰度表,但需要对化合物ID进行标准化(例如,将化合物ID转换为KEGG数据库中的C编号)。
- 输入的表格值:可以是 read counts or normalized values (e.g., TPM, FPKM, RPKM, or relative abundance, corresponds to suitable statistical test method).
- 注意:输入的特征丰度表格不要过滤,以保留所有的背景id信息
metadata:为一个数据框,行为样本,列为样本的表型信息,可以是分类变量也可以是连续变量
注意:metadata的行名与丰度矩阵的列名必须一一对应
下面是一个 KO abundance table 数据示例:
# 示例数据
data("KO_abundance_test")
# 查看前6行
head(KO_abundance[, 1:6])
#> WT1 WT2 WT3 WT4 WT5 WT6
#> K03169 0.002653545 0.005096380 0.002033923 0.000722349 0.003468322 0.001483028
#> K07133 0.000308237 0.000280458 0.000596527 0.000859854 0.000308719 0.000878098
#> K03088 0.002147068 0.002030742 0.003797459 0.004161979 0.002076596 0.003091182
#> K03530 0.003788366 0.000239298 0.000445817 0.000557271 0.000222969 0.000529624
#> K06147 0.000785654 0.001213630 0.001312569 0.001662740 0.002387006 0.001725797
#> K05349 0.001816325 0.002813642 0.003274701 0.001089906 0.002371921 0.001795214
head(metadata)
#> Group Group2
#> WT1 WT G3
#> WT2 WT G3
#> WT3 WT G3
#> WT4 WT G3
#> WT5 WT G3
#> WT6 WT G1
# 查看行列名对应
all(rownames(metadata) %in% colnames(KO_abundance))
## TRUE
intersect(rownames(metadata), colnames(KO_abundance))>0
## TRUE
3、通路数据库
ReporterScore包内置了KEGG通路、模块、基因、化合物和GO(Gene Ontology,基因本体论)数据库,并且还允许使用自定义数据库,这样能够与来自多种组学数据的特征丰度表兼容。
# 1. KEGG pathway-KO and module-KO databases
KOlist <- load_KOlist()
head(KOlist$pathway)
# 2. KEGG pathway-compound and module-compound databases
CPDlist <- load_CPDlist()
head(CPDlist$pathway)
# 3. human (hsa) pathway-ko/gene/compound databases
hsa_pathway_gene <- custom_modulelist_from_org(
org = "hsa",
feature = c("ko", "gene", "compound")[2]
)
head(hsa_pathway_gene)
# 4. GO-gene database
GOlist <- load_GOlist()
head(GOlist$BP)
# 5. 还可以使用下面的函数自定于通路数据库
?custom_modulelist()
4、运行GRSA
使用 GRSA 函数一步运行得到富集结果,其重要参数如下:
- mode:可选 “mixed” 或 “directed”,directed为组间比较分析模式,可以知道通路在哪个组别中显著富集或减少
- method:统计学方法,计算每条通路的pvalue,可选 t.test,wilcox.test、anova、kruskal.test等
- type:选择内置数据库,pathway 或 module 为 KEGG database;‘CC’, ‘MF’, ‘BP’, ‘ALL’为GO数据库
- modulelist:自定义数据库
- feature:特征属性,可以为“ko”, “gene”, “compound”.
接下来,以比较两个组别‘WT-OE’(野生型-过表达)为例子,使用“定向”模式,探索在OE组中哪些通路是富集或减少的。
cat("Comparison: ", levels(factor(metadata$Group)), "\n")
#> Comparison: WT OE
data("genedf")
head(genedf)
数据为一个基因水平的表达矩阵:

GRSA分析:
# Method 1: Set the `feature` and `type`!
reporter_res_gene <- GRSA(genedf,
"Group",
metadata,
mode = "directed",
method = "wilcox.test",
perm = 999,
type = "hsa",
feature = "gene")
## 探索一下结果格式
class(reporter_res_gene)
head(reporter_res_gene)
str(reporter_res_gene)
# view data.frame in Rstudio
View(reporter_res$reporter_s)
# export result as .csv and check using Excel:
export_report_table(reporter_res, dir_name = "./")
生成的结果是一个list对象:
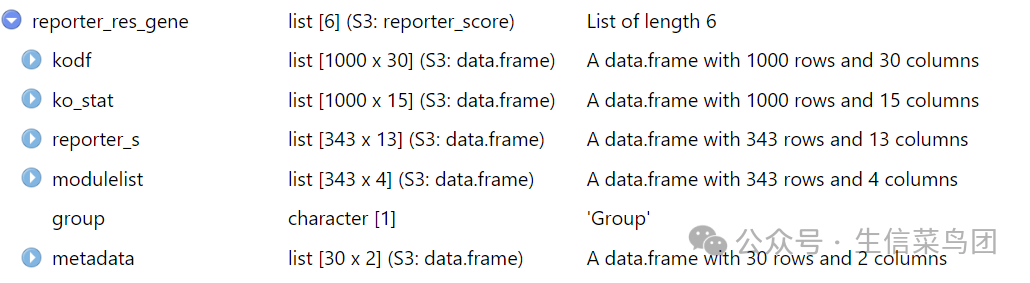
- kodf:输入的 KO_abundance 表格
- ko_stat:ko 统计结果,包括pvalue和z.score
- reporter_s:每条通路的reporter打分
- modulelist:自定义的 modulelist 或者 KOlist数据框
- group:样本分组
- metadata:样本表型metadata数据框
5、结果可视化
得到 reporter 打分后,可以绘制最显著的 富集到的 pathways。
# Use `modify_description` to remove the suffix of pathway description
reporter_res_gene2 <- modify_description(reporter_res_gene, pattern = " - Homo sapiens (human)")
p2 <- plot_report_bar(reporter_res_gene2, rs_threshold = 2)
p2
结果如下:
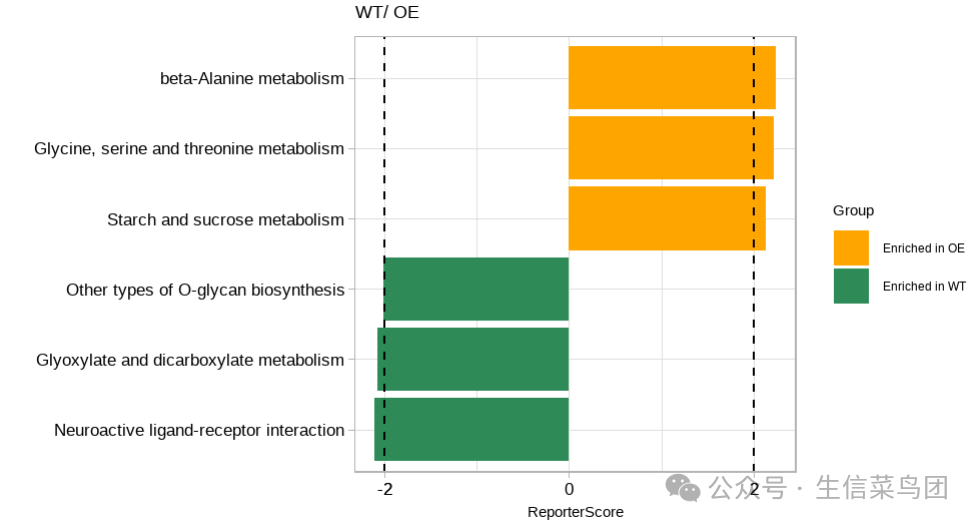
还可以展示KEGG通路的分类:
# View(reporter_res$reporter_s)
plot_report_bar(reporter_res, rs_threshold = c(-2.5, 2.5), facet_level = TRUE)
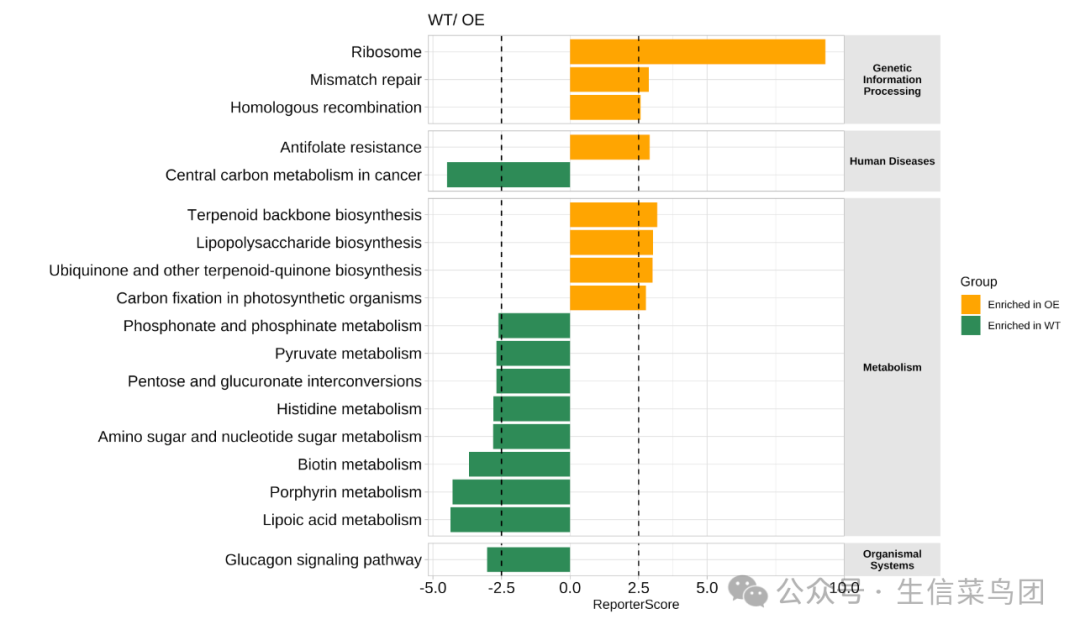
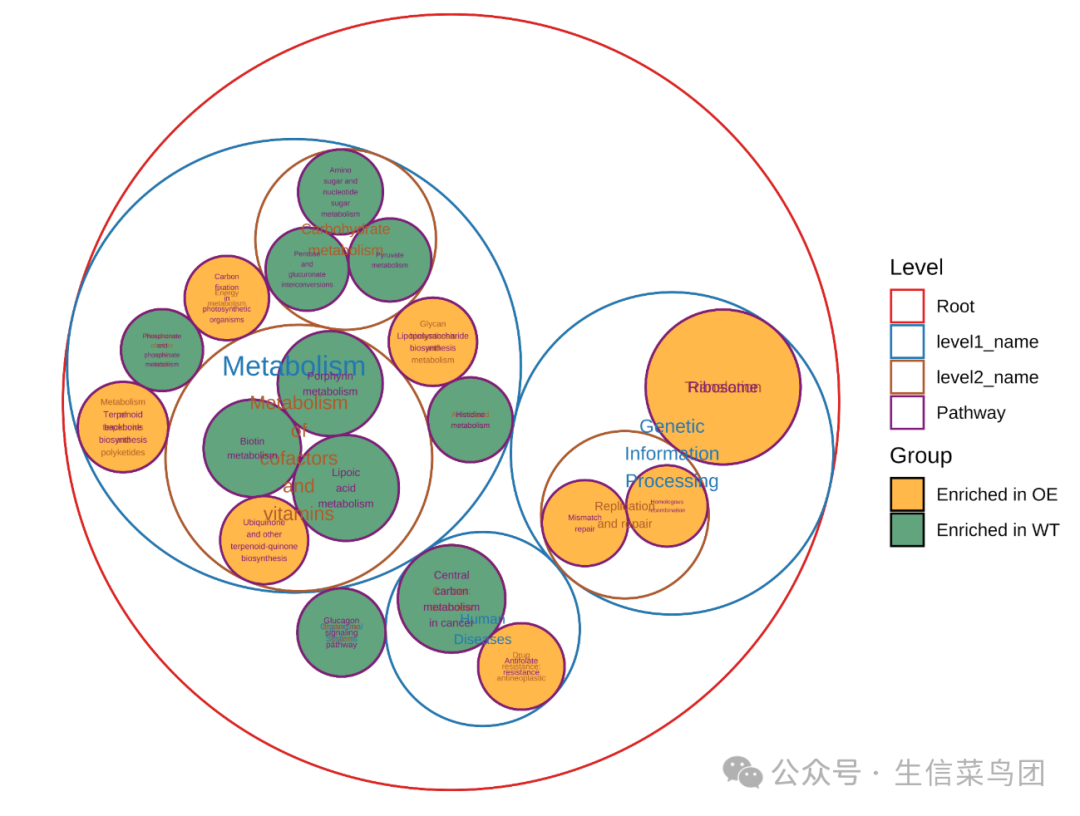
还可以使用圈图风格,这个在代谢和微生物组学中出现的比较多:
plot_report_circle_packing(reporter_res, rs_threshold = c(-2.5, 2.5))
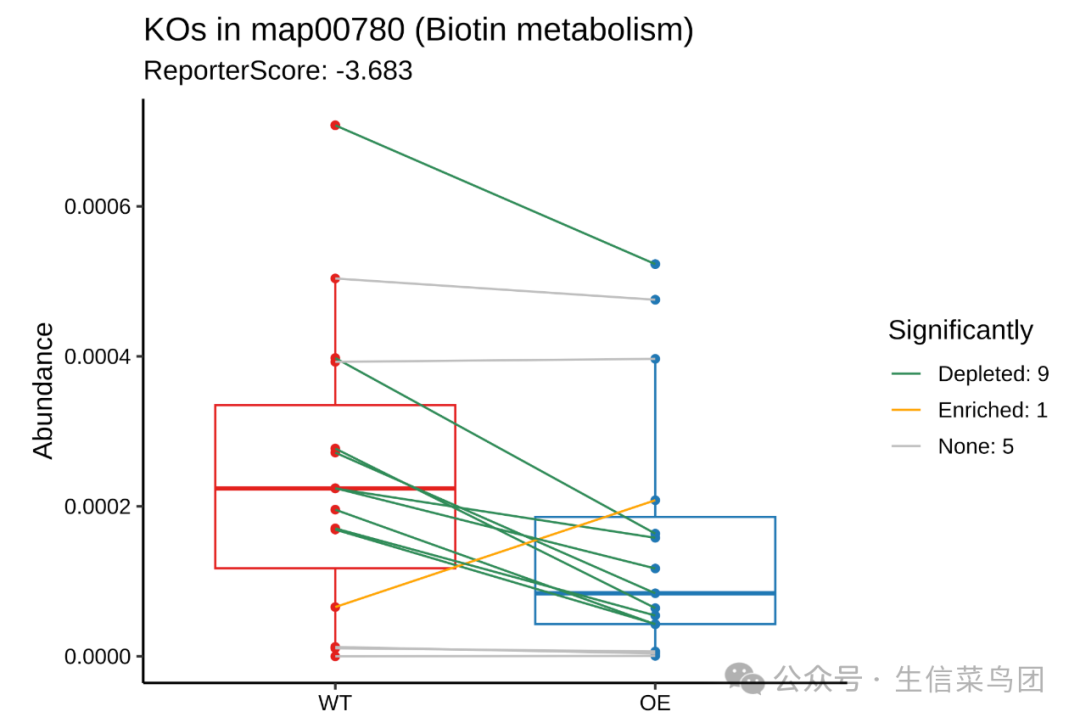
绘制某一条通路的 箱线图:map00780
plot_KOs_in_pathway(reporter_res, map_id = "map00780")
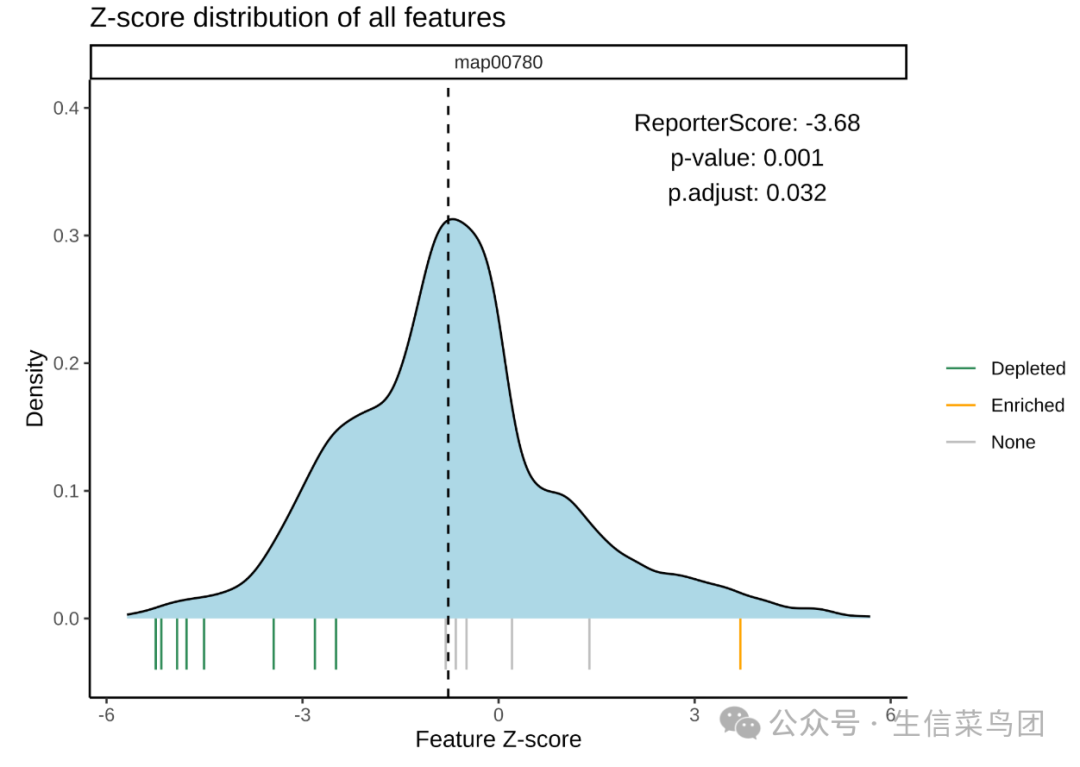
Z-scores分布:
plot_KOs_distribution(reporter_res, map_id = "map00780")
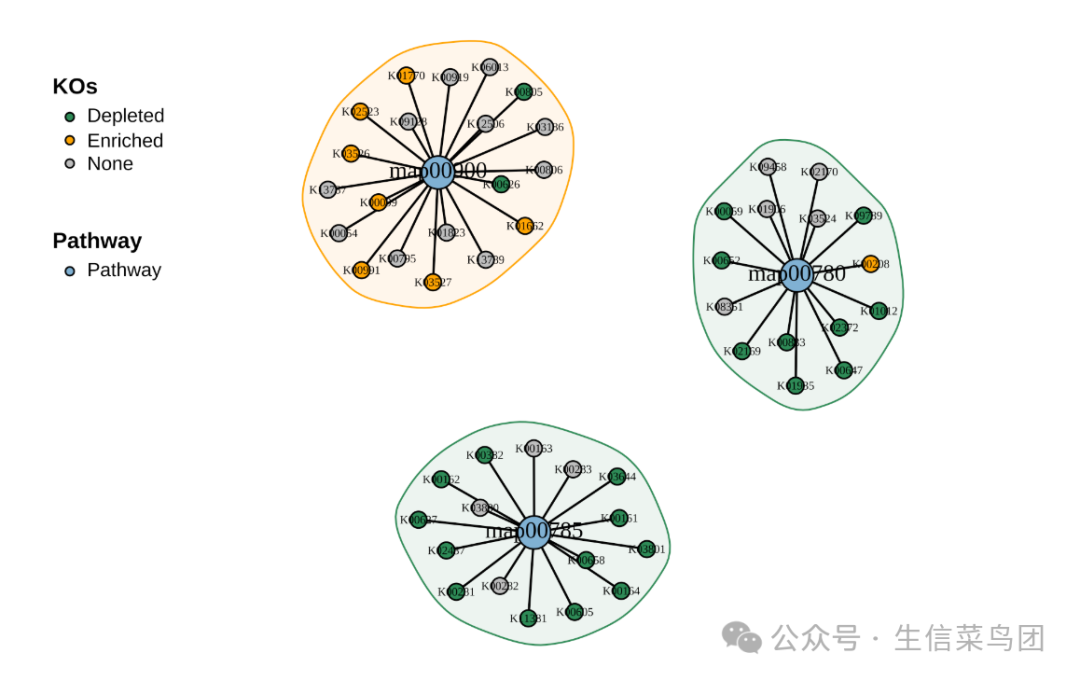
不同通路的网络图:
plot_KOs_network(reporter_res,
map_id = c("map00780", "map00785", "map00900"),
main = "", mark_module = TRUE
)
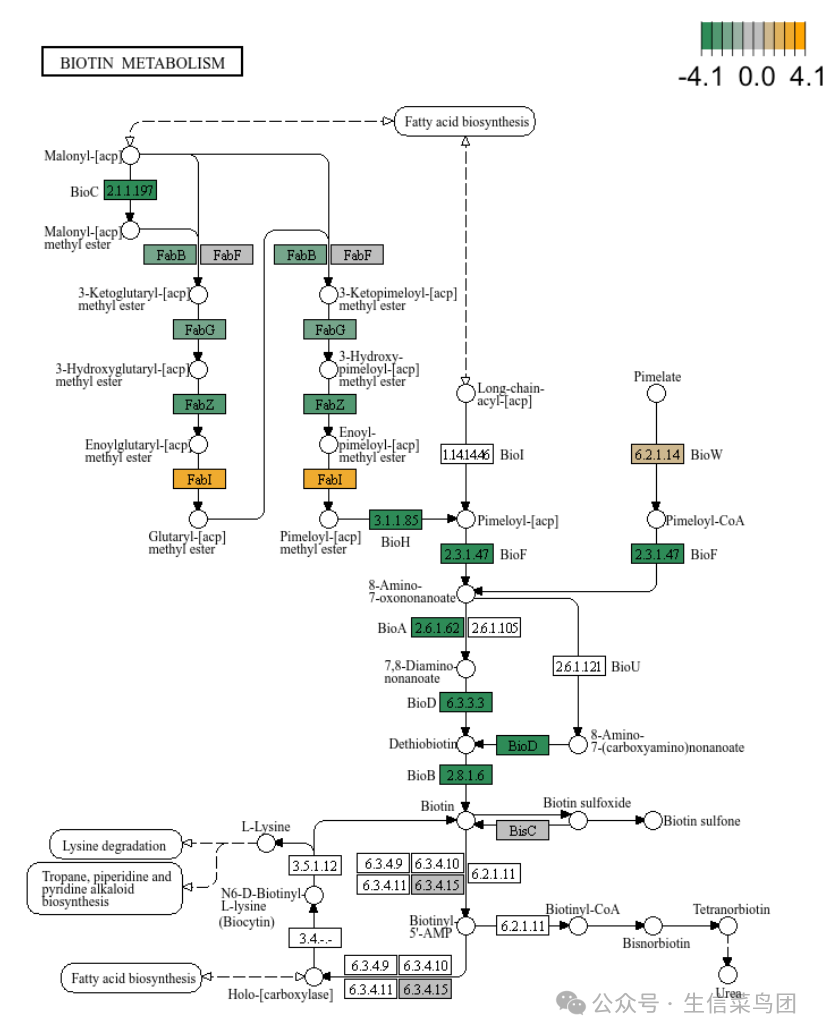
KEGG pathway map图:
plot_KEGG_map(reporter_res, map_id = "map00780", color_var = "Z_score")
其他
还有多分组比较以及其他更多好看的可视化结果,去看看吧~
前面介绍过的其他功能富集分析:
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2025-01-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号