上(市场篇)| 量子计算加速蛋白质折叠


1. 蛋白质折叠是什么
蛋白质折叠(Protein folding)是蛋白质获得其功能性结构和构象的物理过程。通过这一物理过程,蛋白质从无规则卷曲折叠成特定的功能性三维结构。在从mRNA序列翻译成线性的氨基酸链时,蛋白质都是以去折叠多肽或无规则卷曲的形式存在。
蛋白质的基本单位为氨基酸,而蛋白质的一级结构指的就是其氨基酸序列。蛋白质会由所含氨基酸残基的亲水性、疏水性、带正电、带负电等特性通过残基间的相互作用而折叠成一立体的三级结构。
2. 研究蛋白质折叠的目的
因为蛋白质的功能取决于其立体结构,而目前根据已知某基因序列可翻译获得对应蛋白质的氨基酸序列,即蛋白质的一级结构;如果从蛋白质的一级结构就能知道立体结构,那么即可直接从基因推测其编码蛋白质所对应的生物学功能。目前的问题在于,虽然蛋白质可在短时间从一级结构折叠至立体结构,研究者却无法在短时间中从氨基酸序列计算出蛋白质结构,甚至无法得到准确的三维结构。因此,研究蛋白质折叠的过程,可以说是破译折叠密码的过程。
3. “皇冠上的明珠”——蛋白质折叠问题的难度探讨
人体和其他生物体内的蛋白质,都由多种折叠而成。数千个氨基酸组成的长链能自发地折叠成一个稳定的三维结构。理论上讲,使用计算机我们可以推算出一个蛋白质的氨基酸序列折叠后形成的三维结构。因为氨基酸折叠成蛋白质的力学原理很明确,包括氢键、范德华力、疏水作用等相互作用,上千个氨基酸折叠后形成的三维结构,达到了力学最稳态。
不过实际上,蛋白质折叠问题的难度非常大。举个简单的例子,假设每个氨基酸都有2种状态——展开态和折叠态,如果一个蛋白质由100个氨基酸组成,那么它可能的三维结构数量就是2的100次方,这是个非常巨大的天文数字,而其中只有一个结构是稳定的三维结构。
100个氨基酸其实是非常小的蛋白,人体内大多数蛋白质都由数千个氨基酸组成,所以光靠超级计算机的“暴力计算”,是无法根据氨基酸序列预测出蛋白质结构的。这也是蛋白质折叠问题为什么被称为是现代分子生物学的“皇冠上的明珠”。
4. 传统物理和数学方向上的方法和成果
当前传统的研究方法中,诞生了许多分析蛋白质折叠问题的方法,其中效率最高,并最广为人知的就是AlphaFold2折叠。
AlphaFold2是基于AI技术发展而诞生的人工智能系统,由深度思维(DeepMind)公司研发,深度思维就是此前研发出“阿尔法狗”(AlphaGo)的那家公司。AlphaFold2最近在国际蛋白质结构预测大赛中夺冠,它的准确性均分达到了92.4/100,而过去的几十年中,其他传统方法只能在40分左右徘徊。首先让我们来了解一下AlphaFold2的工作原理:
AlphaFold2主要通过预测蛋白质中每对氨基酸之间的距离分布,以及连接它们的化学键之间的角度将所有存在的氨基酸对的测量结果汇总成2D形式的距离直方图,然后让卷积神经网络对这些图片进行学习,从而构建出蛋白质的3D结构。AlphaFold2主要架构如下图:
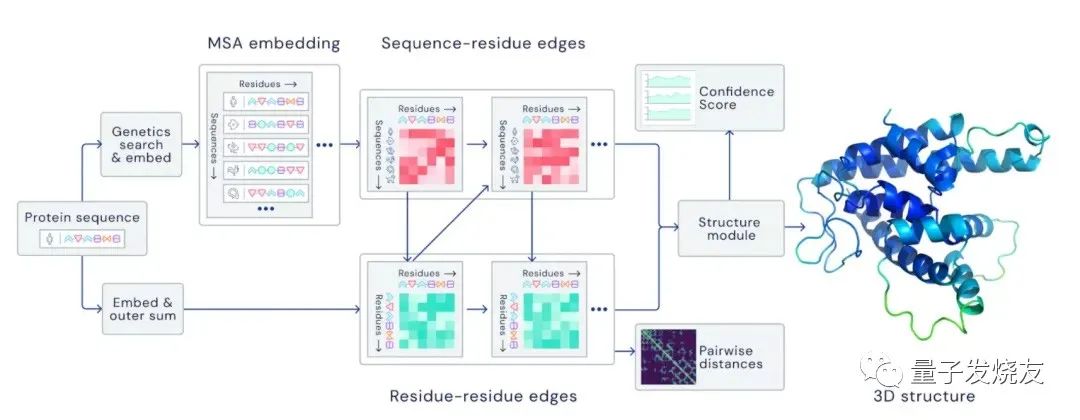
AlphaFold2主要架构
4.1 神经网络EvoFormer
具体来看,AlphaFold2主要利用多序列比对(MSA)把蛋白质的结构和生物信息整合到了深度学习算法中。主要包括两个部分:神经网络EvoFormer和结构模块(Structuremodule)。
在EvoFormer中,主要是将图网络和多序列比对结合完成结构预测。图网络可以很好的表示出事物之间的相关性,它可以将蛋白质的相关信息构建出一个图表,以此表示不同氨基酸之间的距离。
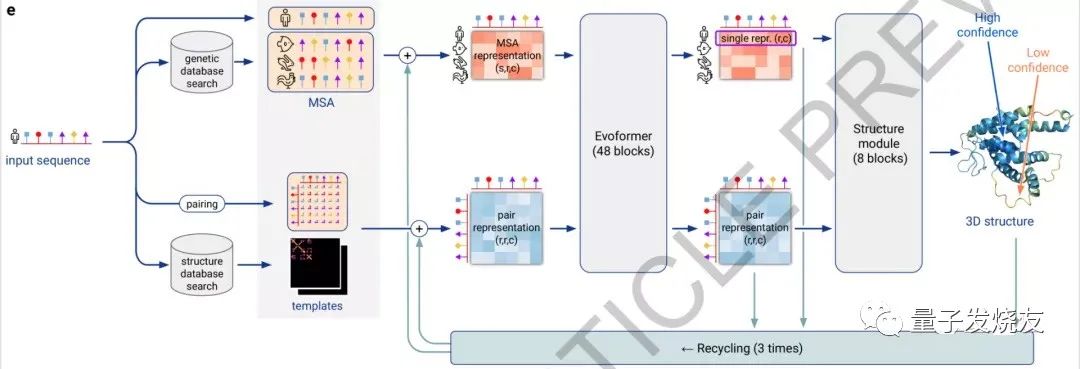
神经网络和结构模块
AlphaFold2的研究人员利用卷积神经网络Attention机制构建出一个特殊的“三重自注意力机制”来处理计算氨基酸之间的关系图,他们会将得到的信息与多序列比对结合。多序列比对主要是使相同的残基位点位于同一列,暴露出不同序列之间的相似部分,从而推断出不同蛋白质在结构和功能上的相似关系。操作方式如下图:
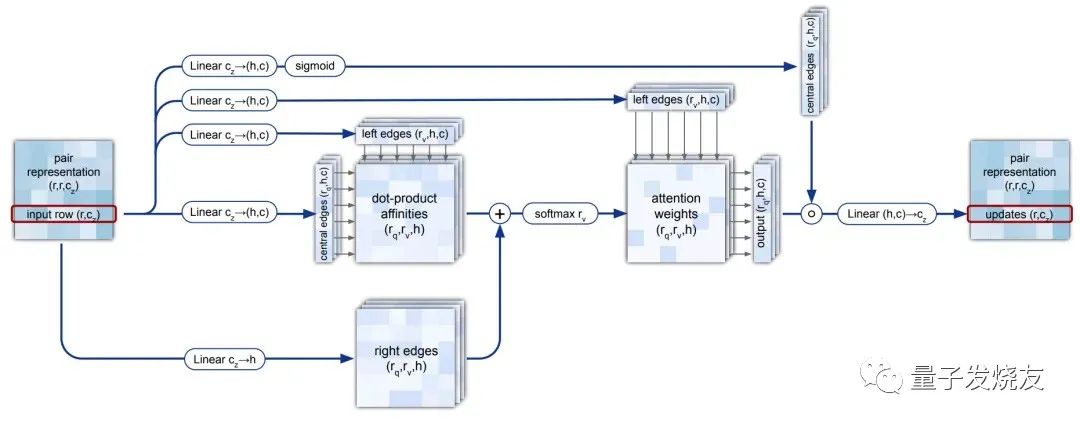
“三重自注意力机制”
4.2 结构模块
结构模块是AlphaFold2架构的第二部分,它的主要工作是将EvoFormer得到的信息转换为蛋白质的3D结构。结构模块工作原理如下:
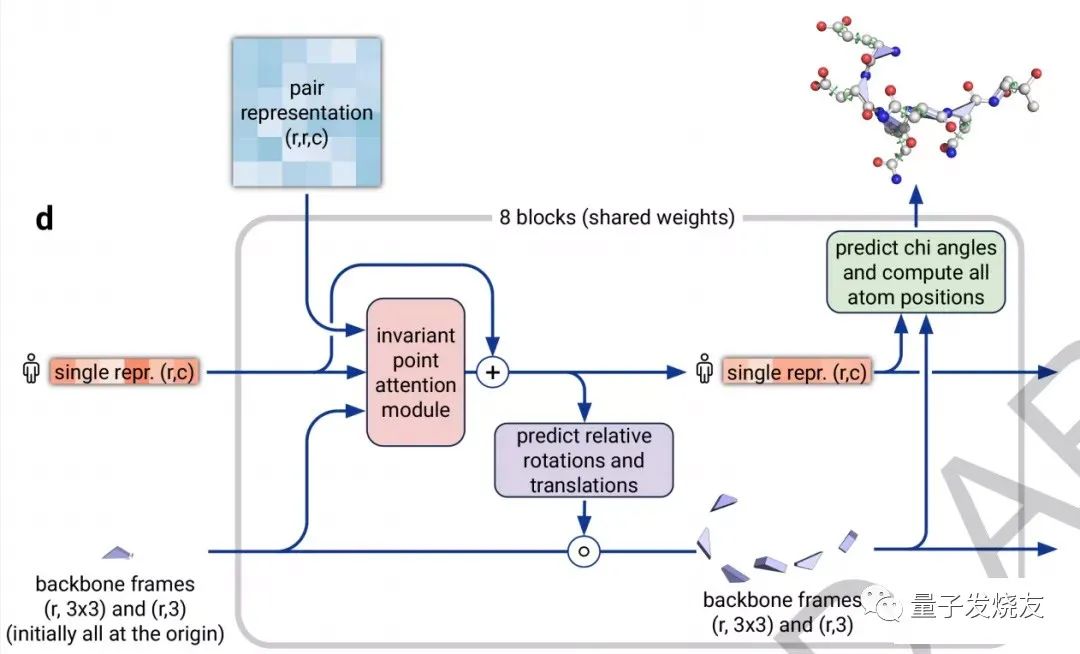
结构模块工作原理
在结构模块中,研究人员同样使用了Attention机制,它可以单独计算蛋白质的各个部分,称为“不变点注意力机制”。是以某个原子为原点,构建出一个3D参考场,根据预测信息进行旋转和平移,得到一个结构框架。具体步骤如下:
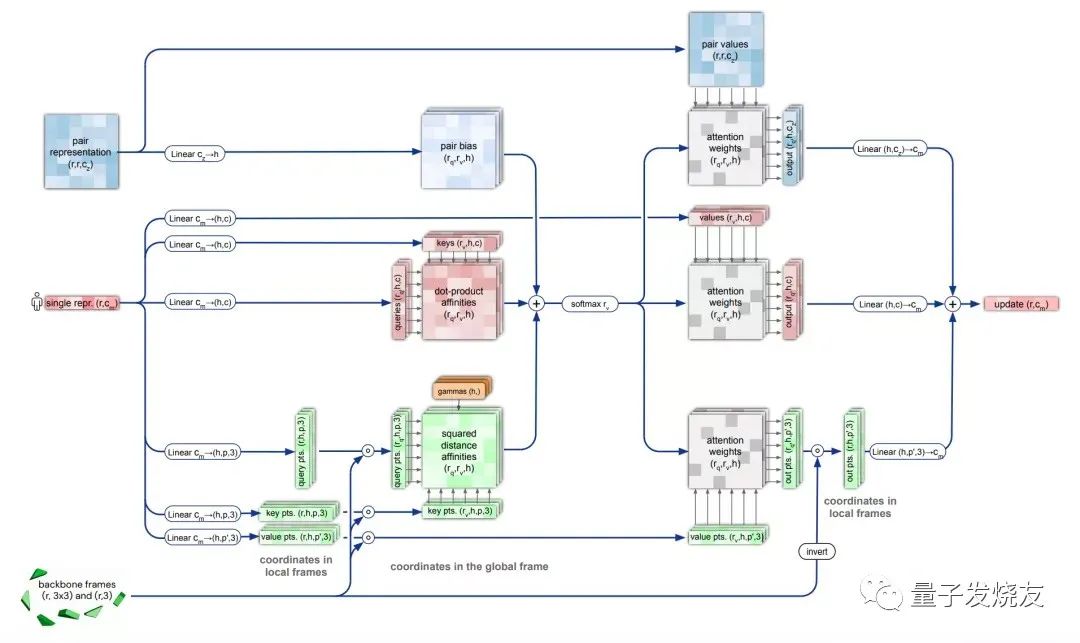
不变点注意力机制
之后Attention机制会对所有原子进行预测,最终汇总得到一个高度准确的蛋白质结构。AlphaFold2的研究人员还强调AlphaFold2是一个"端到端"的神经网络,他们会反复把最终损失应用于输出结果。然后再对输出结果进行递归,不断逼近正确结果,这样做既能减少额外的训练,还能大幅提高预测结构的准确性。
4.3 预测结果
下面我们可以来看看AlphaFold2对于蛋白质折叠预测的效果:
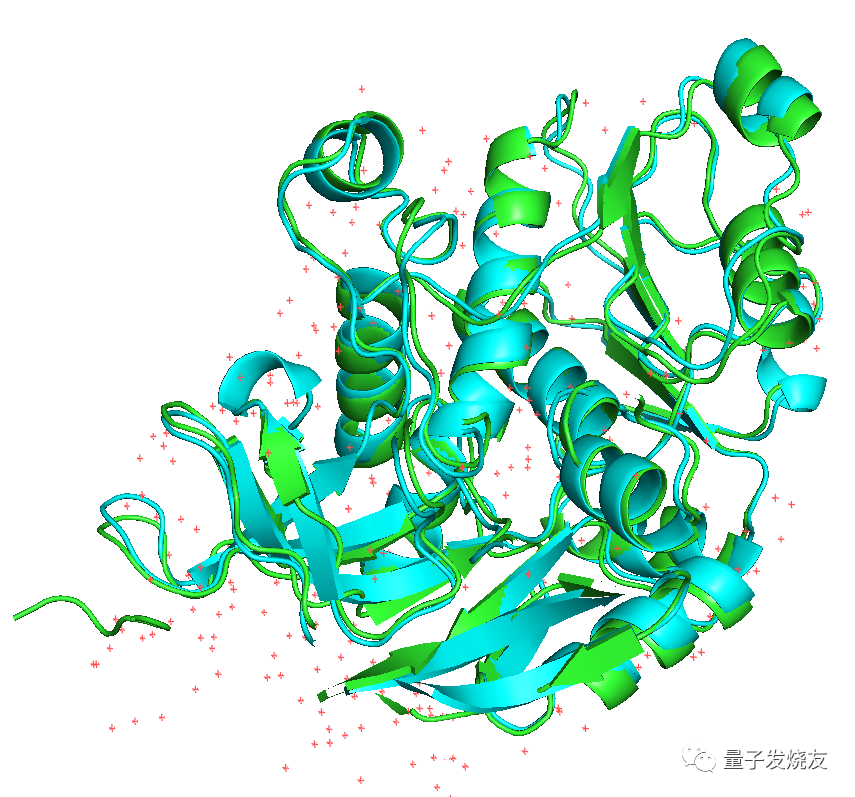
图中显示的是327aa的蛋白与同源结构最高identities = 30%的结构预测结果(青色为预测的,绿色的为解析出来的结构)。两个结构RMSD=0.86埃,预测结果非常好。说明AlphaFold2的预测准确性是很高的。
4.4 AlphaFold2的部分代码
为了对AlphaFold2的运行原理理解更透彻,我们也可以看看它进行蛋白质折叠预测的几个算法运行的例子:
4.4.1 折叠单体
假设有一个具有序列的单体。输入fasta应该是:
>sequence_name
<SEQUENCE>然后运行以下命令:
python3 docker/run_docker.py \
--fasta_paths=monomer.fasta \
--max_template_date=2022-05-10 \
--model_preset=monomer \
--data_dir= $DOWNLOAD_DIR4.4.2. 折叠homomer
假设有一个来自原核生物的同源异构体,它有3个相同序列的拷贝。输入fasta就是:
>sequence_1
<SEQUENCE>
>sequence_2
<SEQUENCE>
>sequence_3
<SEQUENCE>之后运行以下命令:
python3 docker/run_docker.py \
--fasta_paths=homomer.fasta \
--is_prokaryote_list=true \
--max_template_date=2022-05-10 \
--model_preset=multimer \
--data_dir= \$DOWNLOAD_DIR4.4.3. 折叠异聚体
假设有一个未知来源的异聚体A2B3,即具有的2个副本和3个副本,输入fasta:
>sequence_1
<SEQUENCE A>
>sequence_2
<SEQUENCE A>
>sequence_3
<SEQUENCE B>
>sequence_4
<SEQUENCE B>
>sequence_5
<SEQUENCE B>运行以下命令:
python3 docker/run_docker.py \
--fasta_paths=heteromer.fasta \
--is_prokaryote_list=false \
--max_template_date=2022-05-10 \
--model_preset=multimer \
--data_dir= $DOWNLOAD_DIR4.4.4. 一个接一个的折叠多个单体
假设有两个单体,monomer1.fasta和monomer2.fasta。我们可以使用以下命令按顺序折叠两者:
python3 docker/run_docker.py \
--fasta_paths=monomer1.fasta,monomer2.fasta \ --max_template_date=2022-05-10 \
--model_preset=monomer \
--data_dir= $DOWNLOAD_DIR4.4.5. 一个接一个的折叠多个多聚体
假设有两个多聚体,multimer1.fasta和multimer2.fasta。两者都来自原核生物,可以使用以下命令按顺序折叠两者:
python3 docker/run_docker.py \
--fasta_paths=multimer1.fasta,multimer2.fasta \
--is_prokaryote_list=true,true \
--max_template_date=2022-05-10 \
--model_preset=multimer \
--data_dir= $DOWNLOAD_DIR4.4.6. AlphaFold2输出
根据需要预测的氨基酸形式的不同,我们做好每一段的前置代码之后,运行最终的输出程序。输出包括计算的MSA、非松弛结构、松弛结构、排序结构、原始模型输出、预测元数据和部分时间:
<target_name>/
features.pkl
ranked_{0,1,2,3,4}.pdb
ranking_debug.json
relaxed_model_{1,2,3,4,5}.pdb
result_model_{1,2,3,4,5}.pkl
timings.json
unrelaxed_model_{1,2,3,4,5}.pdb
msas/
bfd_uniclust_hits.a3m
mgnify_hits.sto
uniref90_hits.sto5.传统方法的局限性及量子计算的优势
虽然目前以AlphaFold2为代表的人工智能技术,凭借之前通过生物学方法积累的大量蛋白质序列和结构信息,在蛋白质折叠问题上能够对蛋白质结构进行比较精准的预测,但是它们的预测速度还是受到很大的限制,以及无法解决问题内在的NP-hard复杂性和蛋白质序列与结构间高维到高维的映射关系。
而凭借量子态叠加和量子纠缠带来的强大并行计算的能力,量子计算能够加速分析过程,解决上述难题。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2022-08-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号