
使用R创建两个副本的均值的日志散点图。
使用R创建两个副本的均值的日志散点图。
提问于 2018-06-18 02:20:58
我有一些RNA seq数据两种不同的细胞类型(朴素和Th1)。
如何为每个基因(分组变量)找到每种细胞类型的两个副本的平均值,然后绘制这些数据的日志散点图?。
我的桌子是这样的:
df <- data.frame(Gene_name=c("Mrpl15", "Lypla1", "Tcea1"),
naive_A=c(5.21212, 6.62643, 5.74654),
naive_B=c(4.52376, 5.64459, 4.52153),
Th1_A=c(15.50650, 14.46030, 11.57770),
Th1_B=c(5.876490, 5.193010, 2.107200), stringsAsFactors=F)
df
Gene_name naive_A naive_B Th1_A Th1_B
1 Mrpl15 5.21212 4.52376 15.5065 5.87649
2 Lypla1 6.62643 5.64459 14.4603 5.19301
3 Tcea1 5.74654 4.52153 11.5777 2.10720如果有人能帮忙,我会非常感激的!谢谢
回答 1
Stack Overflow用户
发布于 2018-06-18 02:34:33
你可以做这样的事
df %>%
gather(sample, expr, -Gene_name) %>%
mutate(condition = gsub("_\\w$", "", sample)) %>%
group_by(Gene_name, condition) %>%
summarise(expr.mean = mean(expr)) %>%
spread(condition, expr.mean) %>%
ggplot(aes(x = log10(naive), y = log10(Th1), label = Gene_name)) +
geom_point() +
geom_text()
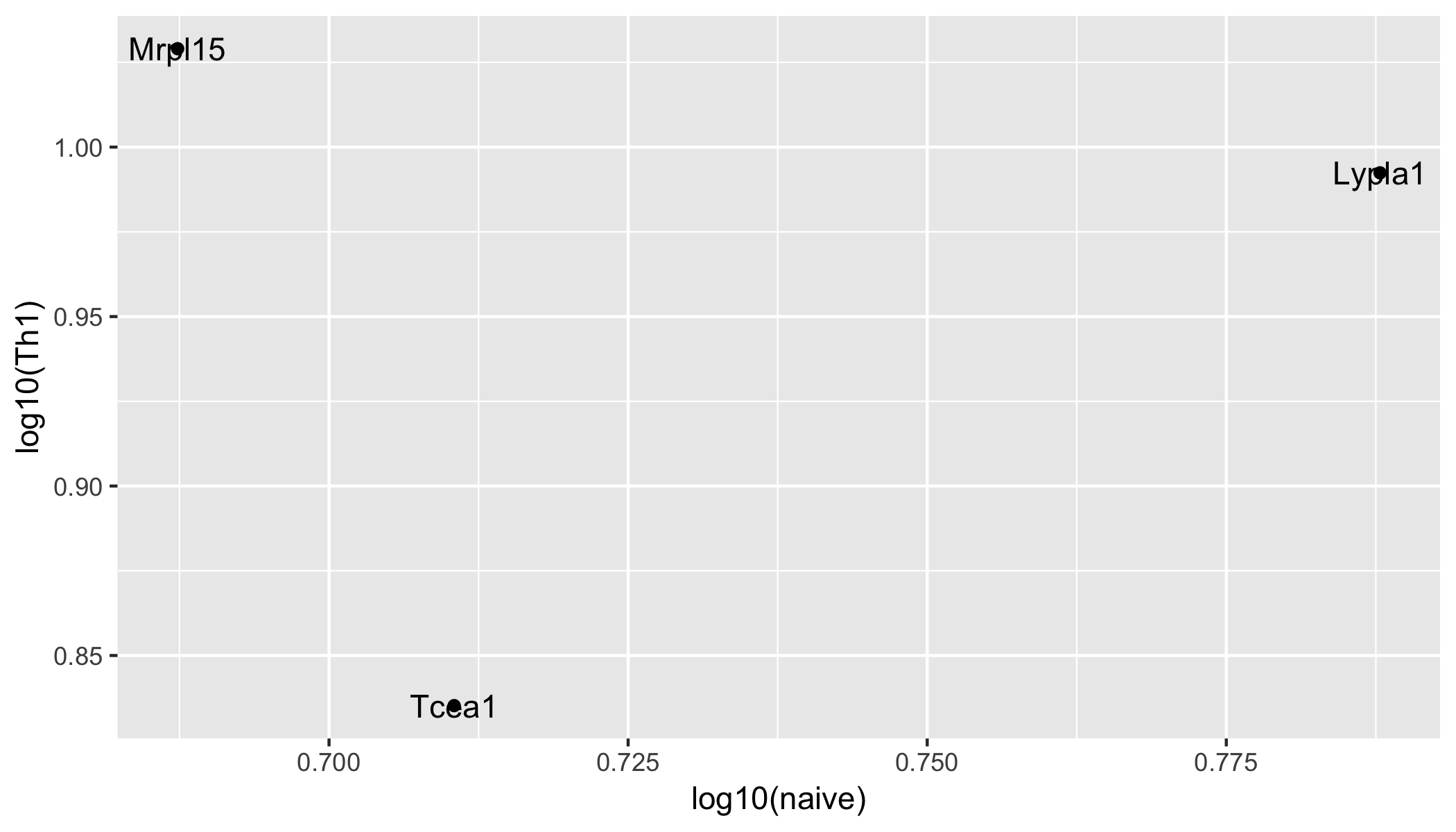
说明:从宽到长的数据,提取一个condition列和每种情况下每个基因的平均表达值。然后从长到宽重新塑造数据,并绘制出从naive到Th1的对数10转换的平均表达量和每个基因的平均表达量。
我只是为了说明而添加了基因标签。您可以通过删除+ geom_text()来删除它们。
样本数据
df <- read.table(text =
"Gene_name naive_A naive_B Th1_A Th1_B
1 Mrpl15 5.21212 4.52376 15.50650 5.876490
2 Lypla1 6.62643 5.64459 14.46030 5.193010
3 Tcea1 5.74654 4.52153 11.57770 2.107200", header = T)页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/50901980
复制相关文章
相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号