
R:在现有的dataframe中展开数据格式列
R:在现有的dataframe中展开数据格式列
提问于 2017-03-28 21:46:30
我正在使用jsonlite::fromJSON将一个JSON文档获取到一个dataframe。问题是,它没有被完全夷为平地--原因我不知道(但下面有人提到过,因为他们是以字典的形式出现的,他们不会被夷为平地)。
df <- jsonlite::fromJSON(out, flatten = TRUE, simplifyDataFrame = TRUE)
n <- df$hits$total # 1 entry
dat <- df$hits$hits
# dat is a dataframe
dim(dat)
[1] 2 5
# class of each column in the dataframe
lapply(dat, class)
$`_index`
[1] "character"
$`_type`
[1] "character"
$`_id`
[1] "character"
$`_score`
[1] "numeric"
$`_source.samples`
[1] "list"有一个列dat$_source.samples,它实际上是一个数据格式列表:
> dim(dat$`_source.samples`[[1]])
[1] 21 2
> dim(dat$`_source.samples`[[2]])
[1] 21 2如何展开包含dataframes的列,使它们在现有的dataframe中形成两个新列,并复制前四行作为展开的结果。下面是一个示例:
dat外观的Rstudio屏幕截图:
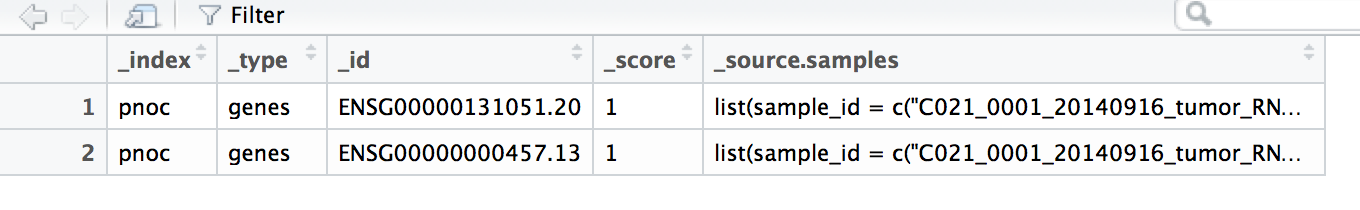
# the first four columns look like this
> head(dat[,1:4])
_index _type _id _score
1 pnoc genes ENSG00000131051.20 1
2 pnoc genes ENSG00000000457.13 1
# the fifth column `_source.samples` that has dataframes looks like this
# just showing the dataframe in the first row of `dat`
> head(dat$`_source.samples`[[1]])
sample_id rsem.fpkm
1 C021_0001_20140916_tumor_RNASeq 39.11
2 CPBT_0001_1_tumor_RNASeq 184.56
3 CPBT_0007_1_tumor_RNASeq 41.29
4 C021_0010_001774_tumor_RNASeq 86.31
5 C021_0003_001409_tumor_RNASeq 79.24
6 CPBT_0005_1_tumor_RNASeq 66.20所以我想要这样的东西:
_index _type _id _score sample_id
1 pnoc genes ENSG00000000457.13 1 C021_0001_20140916_tumor_RNASeq
2 pnoc genes ENSG00000000457.13 1 CPBT_0001_1_tumor_RNASeq
3 pnoc genes ENSG00000000457.13 1 CPBT_0007_1_tumor_RNASeq
4 pnoc genes ENSG00000000457.13 1 C021_0010_001774_tumor_RNASeq
5 pnoc genes ENSG00000000457.13 1 C021_0003_001409_tumor_RNASeq
6 pnoc genes ENSG00000000457.13 1 CPBT_0005_1_tumor_RNASeq
rsem.fpkm
1 1.39
2 5.58
3 1.93
4 3.64
5 5.20
6 3.69下面是一个可复制的数据集:
> dput(dat)
structure(list(`_index` = c("pnoc", "pnoc"), `_type` = c("genes",
"genes"), `_id` = c("ENSG00000131051.20", "ENSG00000000457.13"
), `_score` = c(1, 1), `_source.samples` = list(structure(list(
sample_id = c("C021_0001_20140916_tumor_RNASeq", "CPBT_0001_1_tumor_RNASeq",
"CPBT_0007_1_tumor_RNASeq", "C021_0010_001774_tumor_RNASeq",
"C021_0003_001409_tumor_RNASeq", "CPBT_0005_1_tumor_RNASeq",
"CPBT_0008_1_tumor_RNASeq", "C021_0002_001113_tumor_RNASeq",
"C021_0013_001872_tumor_RNASeq", "C021_0005_001661_tumor_RNASeq",
"C021_0007_001669_tumor_RNASeq", "C021_0008_001699_tumor_RNASeq",
"CPBT_0006_1_tumor_RNASeq", "C021_0011_001786_tumor_RNASeq",
"C021_0009_001766_tumor_RNASeq", "CPBT_0004_1_tumor_RNASeq",
"CPBT_0003_1_tumor_RNASeq", "CPBT_0009_1_tumor_RNASeq", "C021_0006_001666_tumor_RNASeq",
"C021_0012_001825_tumor_RNASeq", "C021_0004_001418_tumor_RNASeq"
), rsem.fpkm = c(39.11, 184.56, 41.29, 86.31, 79.24, 66.2,
42.13, 88.78, 78.73, 96.79, 38.5, 105.12, 129.16, 145.13,
117.96, 86.53, 75.43, 179.01, 0, 61.61, 98.64)), .Names = c("sample_id",
"rsem.fpkm"), class = "data.frame", row.names = c(NA, 21L)),
structure(list(sample_id = c("C021_0001_20140916_tumor_RNASeq",
"CPBT_0001_1_tumor_RNASeq", "CPBT_0007_1_tumor_RNASeq", "C021_0010_001774_tumor_RNASeq",
"C021_0003_001409_tumor_RNASeq", "CPBT_0005_1_tumor_RNASeq",
"CPBT_0008_1_tumor_RNASeq", "C021_0002_001113_tumor_RNASeq",
"C021_0013_001872_tumor_RNASeq", "C021_0005_001661_tumor_RNASeq",
"C021_0007_001669_tumor_RNASeq", "C021_0008_001699_tumor_RNASeq",
"CPBT_0006_1_tumor_RNASeq", "C021_0011_001786_tumor_RNASeq",
"C021_0009_001766_tumor_RNASeq", "CPBT_0004_1_tumor_RNASeq",
"CPBT_0003_1_tumor_RNASeq", "CPBT_0009_1_tumor_RNASeq", "C021_0006_001666_tumor_RNASeq",
"C021_0012_001825_tumor_RNASeq", "C021_0004_001418_tumor_RNASeq"
), rsem.fpkm = c(1.39, 5.58, 1.93, 3.64, 5.2, 3.69, 1.75,
5.38, 3.46, 4.14, 0.96, 3.93, 4.47, 3.17, 4.38, 2.8, 2.27,
7.4, 0, 2.76, 5.55)), .Names = c("sample_id", "rsem.fpkm"
), class = "data.frame", row.names = c(NA, 21L)))), .Names = c("_index",
"_type", "_id", "_score", "_source.samples"), class = "data.frame", row.names = 1:2)谢谢!
回答 1
Stack Overflow用户
回答已采纳
发布于 2017-03-28 21:55:34
这些列不会被扁平化,因为它们最初是在json文件中嵌入的字典。
下面是你可以做的,尽管这并不是完全直观的:
首先,使用列_source.samples的内容创建一个新的dataframe。您需要确保保留_id列,以确定每个数据来自哪一行。
samples <- mapply(function(x, y) cbind(x, _id = y), dat$`_source.samples, dat$_id, SIMPLIFY = FALSE)然后将其与dat合并。
merge(dat, rbind.pages(samples), by= "_id")页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/43080531
复制相关文章
相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号