
计算每个单元格中给定字符的出现次数
问题
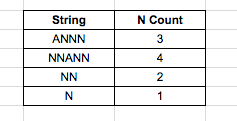
例如,如果我想在一列字符串中计算N的数量,如何在每个单元格的基础上在谷歌电子表格中这样做(也就是说,一个公式每次指向一个单元格,而我可以向下拖放)?
背景
我必须为一个名为汤姆姆**的程序确定一个阈值汤姆姆,它比较小DNA母题*的PWMs*之间的相似性,N是字母A、C、G和T的任何线性组合的正则表达式。如果我能知道我的DNA基元的非N长度的分布,以帮助我知道TOMTOM的适当-min-overlap <integer>值,那就太好了。
下面是一些真实的例子:
** TOMTOM是一个将DNA主题与已知主题数据库进行比较的工具。有关更多信息,请参见这里。
* PWM代表位置权重矩阵:
- 根据Wiki:位置权重矩阵(PWM),也称为位置特定权重矩阵(PSSM),是生物序列中常用的模式表示。
- 根据本文,它可以定义为:
位置权重矩阵(PWM)或类PWM模型被广泛用于表达蛋白质的DNA结合偏好(Stormo,2000)。在这些模型中,使用一个矩阵来表示TF结合位点(TFBS),每个元素代表来自相应位置的核苷酸对整个结合亲和力的贡献。传统PWM模型的一个固有假设是位置无关,即假设TFBS中不同核苷酸位置对整个结合亲和力的贡献是加性的。虽然这种近似是广泛有效的,但它不适用于几种蛋白质(Man & Stormo,2001年;Bulyk等人,2002年)。为了改进定量建模,将PWM模型扩展到包括其他参数,如k-mer特征,以说明TFBSs中的位置依赖关系(赵等人,2012年;Mathelier & Wasserman,2013年;Mordelet al,2013年;Weirauch等人,2013年;Riley等人,2015年)。核苷酸位置之间的相互依赖有一个结构来源。例如,相邻碱基对之间的叠加作用形成了局部的三维DNA结构.TFs对序列依赖的DNA构象有偏好,我们称之为DNA形状读出(Rohs等人,2009,2010)。
或,更多的同时代:
基于这一理论基础,增加传统PWM模型的另一种方法是纳入DNA结构特征。结合这些DNA形状特征的TF-DNA结合特异性模型取得了与包含高阶k-mer特征的模型相当的性能水平,同时需要更少的参数(周等人,2015年)。我们先前揭示了DNA形状读出对基本螺旋环螺旋(bHLH)和同源结构域TF家族成员的重要性(Dror等人,2014年;Yang等人,2014年;周等人,2015年)。对于Hox TFs,我们还能够确定TFBS中的哪些区域使用DNA形状读出,显示了该方法揭示TF-DNA识别的机械洞察力的力量(Abe等人,2015年)。由于缺乏大规模的高质量的TF-DNA结合数据,这种能力仅在两个蛋白家族中得到了广泛的显示。随着最近大量的蛋白质- DNA结合的高通量测量,现在有可能剖析DNA形状读出在许多TF家族中的作用。
* DNA基序:维基:在遗传学中,序列基序是一种广泛存在并具有或推测具有生物学意义的核苷酸或氨基酸序列模式。对于蛋白质来说,序列基序与结构基序是不同的,结构模体是由氨基酸的三维排列而形成的,而氨基酸可能不是相邻的。
回答 3
Stack Overflow用户
发布于 2014-11-27 00:39:40
一次一个单元格的替代方案(要复制的公式):
=len(A2)-len(SUBSTITUTE(A2,"N",""))Stack Overflow用户
发布于 2014-11-26 19:51:36
我不知道这是否有帮助,但假设你在A2:A6范围内有这些字符串,然后输入
=ArrayFormula(LEN(REGEXREPLACE(A2:A6, "[^N]", "")))在B2中,应该输出整个范围的N计数。
Stack Overflow用户
发布于 2018-10-06 17:25:05
=len(A2)-len(SUBSTITUTE(A2,"N",""))这是可行的,但是如果您想找到与特定模式匹配的所有数字,比如说3。
=len(A2)-len(SUBSTITUTE(A2,"3",""))是你所需要的。
https://stackoverflow.com/questions/27154682
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号