
重叠两个基因集,找出它们的重叠意义并绘制它们
(图3a,b,扩展数据图3a,b及补充表1)。48h后,1/3以上的转录组差异表达(>5,000个基因;405个编码胞外区蛋白质的基因,基因本体(GO)登录0005576),与维穆拉非尼治疗5天后体内A375肿瘤的基因表达变化明显重叠(图3a,b和扩展数据图3c)。Colo800和UACC62黑色素瘤细胞在维母拉非尼和H3122肺腺癌细胞中也有类似的广泛表达。尽管有不同的细胞来源、不同的致癌因素和不同的靶向治疗,但黑色素瘤分泌体与肺腺癌细胞之间仍有明显的重叠(P < 9.11×10−5)。
原始纸
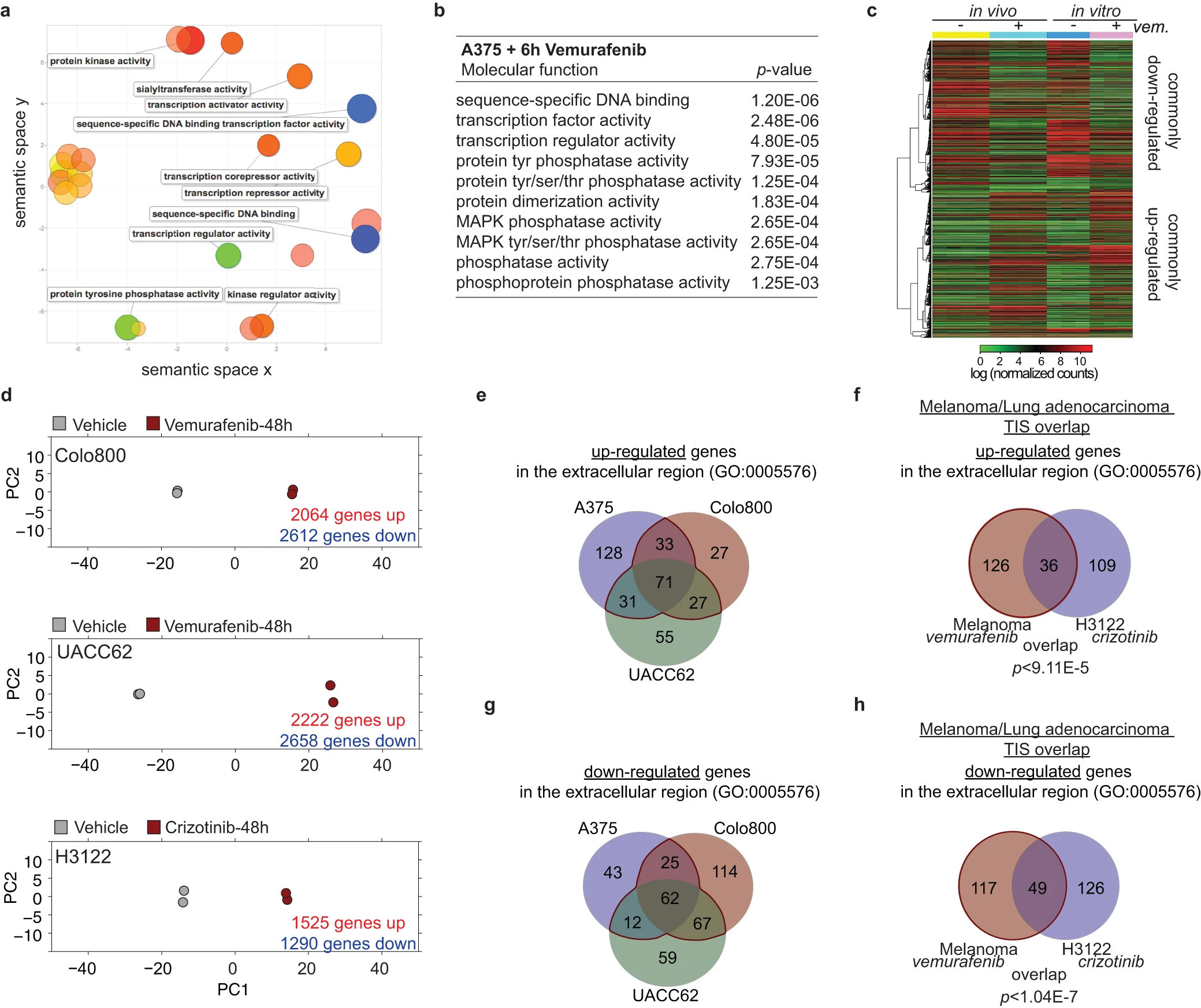
我希望看到类似于图f,其中它显示了交集和意义重叠。为了达到这个目的,我让这段代码一直工作到交集部分,但我不知道如何运行这个重要部分。
library(reshape2)
library(venneuler)
RNA_seq_cds <- read.csv("~/Downloads/RNA_seq_gene_set.txt", header=TRUE, sep="\t")
head(RNA_seq_cds)
ATAC_seq <- read.csv("~/Downloads/ATAC_seq_gene_set.txt", header=TRUE, sep="\t")
head(ATAC_seq)
RNA_seq <- RNA_seq_cds
ATAC_seq <- ATAC_seq
#https://stackoverflow.com/questions/6988184/combining-two-data-frames-of-different-lengths
cbindPad <- function(...) {
args <- list(...)
n <- sapply(args, nrow)
mx <- max(n)
pad <- function(x, mx) {
if (nrow(x) < mx) {
nms <- colnames(x)
padTemp <- matrix(NA, mx - nrow(x), ncol(x))
colnames(padTemp) <- nms
if (ncol(x) == 0) {
return(padTemp)
} else {
return(rbind(x, padTemp))
}
} else {
return(x)
}
}
rs <- lapply(args, pad, mx)
return(do.call(cbind, rs))
}
dat <- cbindPad(ATAC_seq, RNA_seq)
vennfun <- function(x) {
x$id <- seq(1, nrow(x)) #add a column of numbers (required for melt)
xm <- melt(x, id.vars="id", na.rm=TRUE) #melt table into two columns (value & variable)
xc <- dcast(xm, value~variable, fun.aggregate=length) #remove NA's, list presence/absence of each value for each variable (1 or 0)
rownames(xc) <- xc$value #value column=rownames (required for Venneuler)
xc$value <- NULL #remove redundent value column
xc #output the new dataframe
}
#https://stackoverflow.com/questions/9121956/legend-venn-diagram-in-venneuler
VennDat <- vennfun(dat)
genes.venn <- venneuler(VennDat)
genes.venn$labels <- c("RNA", "\nATAC" )
# plot(genes.venn, cex =15, )
#https://stackoverflow.com/questions/30225151/how-to-create-venn-diagram-in-r-studio-from-group-of-three-frequency-column
#https://rstudio-pubs-static.s3.amazonaws.com/13301_6641d73cfac741a59c0a851feb99e98b.html
vd <- venneuler(VennDat)
vd$labels <- paste(genes.venn$labels, colSums(VennDat))
plot(vd, cex=10)
text(.3, .45,
bquote(bold("Common ="~.(as.character(sum(rowSums(VennDat) == 2))))),
col="red", cex=1)
LABS <- vd$labels上面的代码给出了相交图。
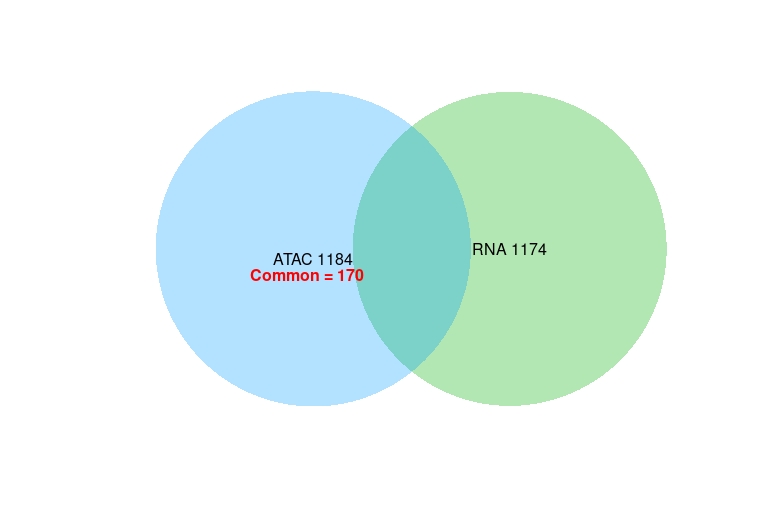
现在,重要意义部分,我如何在两个基因集之间这样做,并显示它,如原始图所示。
我的数据,我用来生成上面的图
任何建议或帮助都将不胜感激。
回答 1
Stack Overflow用户
发布于 2021-09-11 17:50:28
如果你谈论如何在你的数字下放置任何文本,就像你以前做的那样使用“文本”。只是猜测一下x=和y=的坐标。xpd=TRUE允许您在边距上进行绘图。
VennDat <- vennfun(dat)
vd <- venneuler(VennDat)
vd$labels <- paste(c("RNA", "ATAC"), colSums(VennDat))
plot(vd, cex=10, border=c(NA, 'red'), col=c('#6b65af', '#ad7261'))
text(x=.5, y=.5, sum(rowSums(VennDat) == 2), xpd=TRUE)
text(.5, .15, 'overlap\n', xpd=TRUE)
text(.5, .13, bquote(italic(p)*'< 9.11E-55'), xpd=TRUE)
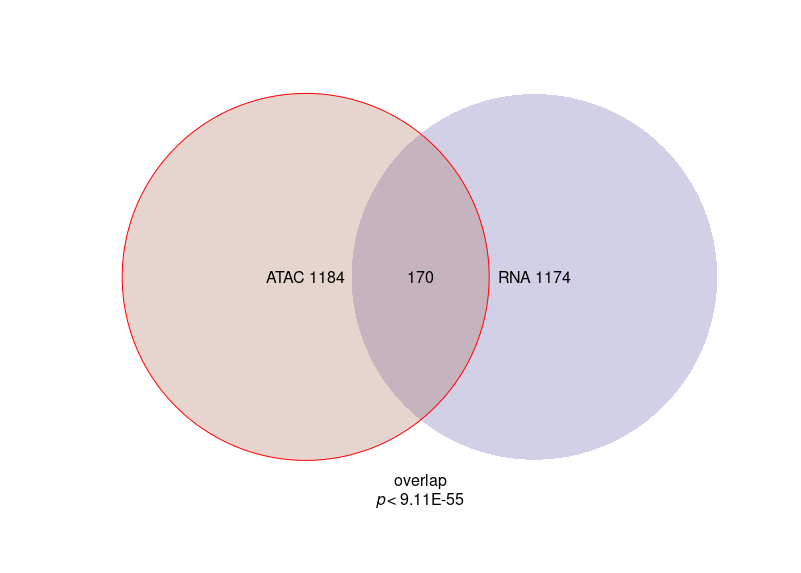
我还调整了plot的一些参数。您可以使用以下方法检查绘图方法的代码:
venneuler:::plot.VennDiagram如果你想知道重要性是如何计算出来的,你应该在交叉验证上发布你的问题。
https://stackoverflow.com/questions/69143816
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号