
在R中获取两个文件之间的交集
在R中获取两个文件之间的交集
提问于 2020-12-16 21:51:04
我有人类外显子的位置(染色体数目,外显子开始和外显子末端)
> head(a1)
exon Chromosome Start End
1 uc001aaa.3 1 11873 12227
2 uc001aaa.3 1 12612 12721
3 uc001aaa.3 1 13220 14409
4 uc010nxr.1 1 11873 12227
5 uc010nxr.1 1 12645 12697
6 uc010nxr.1 1 13220 14409
> str(a)
'data.frame': 8 obs. of 4 variables:
$ exon : chr "uc001aaa.3" "uc001aaa.3" "uc001aaa.3" "uc010nxr.1" ...
$ Chromosome: int 1 1 1 1 1 1 1 1
$ Start : int 11873 12612 13220 11873 12645 13220 11873 12594
$ End : int 12227 12721 14409 12227 12697 14409 12227 12721
> dput(a)
structure(list(exon = c("uc001aaa.3", "uc001aaa.3", "uc001aaa.3",
"uc010nxr.1", "uc010nxr.1", "uc010nxr.1", "uc010nxq.1", "uc010nxq.1"
), Chromosome = c(1L, 1L, 1L, 1L, 1L, 1L, 1L, 1L), Start = c(11873L,
12612L, 13220L, 11873L, 12645L, 13220L, 11873L, 12594L), End = c(12227L,
12721L, 14409L, 12227L, 12697L, 14409L, 12227L, 12721L)), class = "data.frame", row.names = c(NA,
-8L))
>我有另一份文件
> head(a2)
SampleID Chromosome Start End
1 sampel1 1 64613 5707515
2 sampel1 1 5712940 5732322
3 sampel1 1 5732399 16383682
4 sampel1 1 16383742 16389288
5 sampel1 1 16390813 16830026
6 sampel1 1 16830201 17278112
> str(a2)
'data.frame': 7 obs. of 4 variables:
$ SampleID : chr "sampel1" "sampe1" "sampel1" "sampel1" ...
$ Chromosome: int 1 1 1 1 1 1 1
$ Start : int 64613 5712940 5732399 16383742 16390813 16830201 17284498
$ End : int 5707515 5732322 16383682 16389288 16830026 17278112 120374803
> dput(a2)
structure(list(SampleID = c("sampel1", "sampe1", "sampel1", "sampel1",
"sampel1", "sampel1", "sampel1"), Chromosome = c(1L, 1L, 1L,
1L, 1L, 1L, 1L), Start = c(64613L, 5712940L, 5732399L, 16383742L,
16390813L, 16830201L, 17284498L), End = c(5707515L, 5732322L,
16383682L, 16389288L, 16830026L, 17278112L, 120374803L)), class = "data.frame", row.names = c(NA,
-7L))
>我想知道在第二个文件的间隔中有多少外显子。
在第二个文件中,假设64613到5707515中有多少外显子
我的欲望输出就像
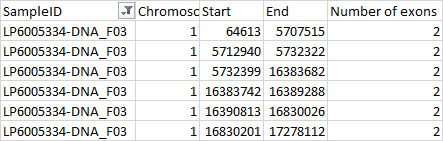
回答 1
Stack Overflow用户
发布于 2020-12-16 22:01:28
您正在寻找GenomicRanges包:
library(GenomicRanges)
a1.GRanges <- GRanges(a1$Chromosome,
ranges = IRanges(a1$Start, a1$End),
seqinfo = a1$exons)
a2.GRanges <- GRanges(a2$Chromosome,
ranges = IRanges(a2$Start, a2$End),
seqinfo = a2$SampleID)
findOverlaps(a2.GRanges,a1.GRanges)正如@A5C1D2H2I1M1N2O1R2T1所暗示的那样,在这些范围内没有重叠。
页面原文内容由Stack Overflow提供。腾讯云小微IT领域专用引擎提供翻译支持
原文链接:
https://stackoverflow.com/questions/65331625
复制相关文章
相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号