
regex匹配不工作于简单字符串与Pyteomics解析器
我正在对人类蛋白质组进行硅化消化,这意味着我试图在某一特定位置切割每一种蛋白质的氨基酸序列。我在我创建的一个更大的函数中使用Pyteomics解析器函数派提组学分析者。
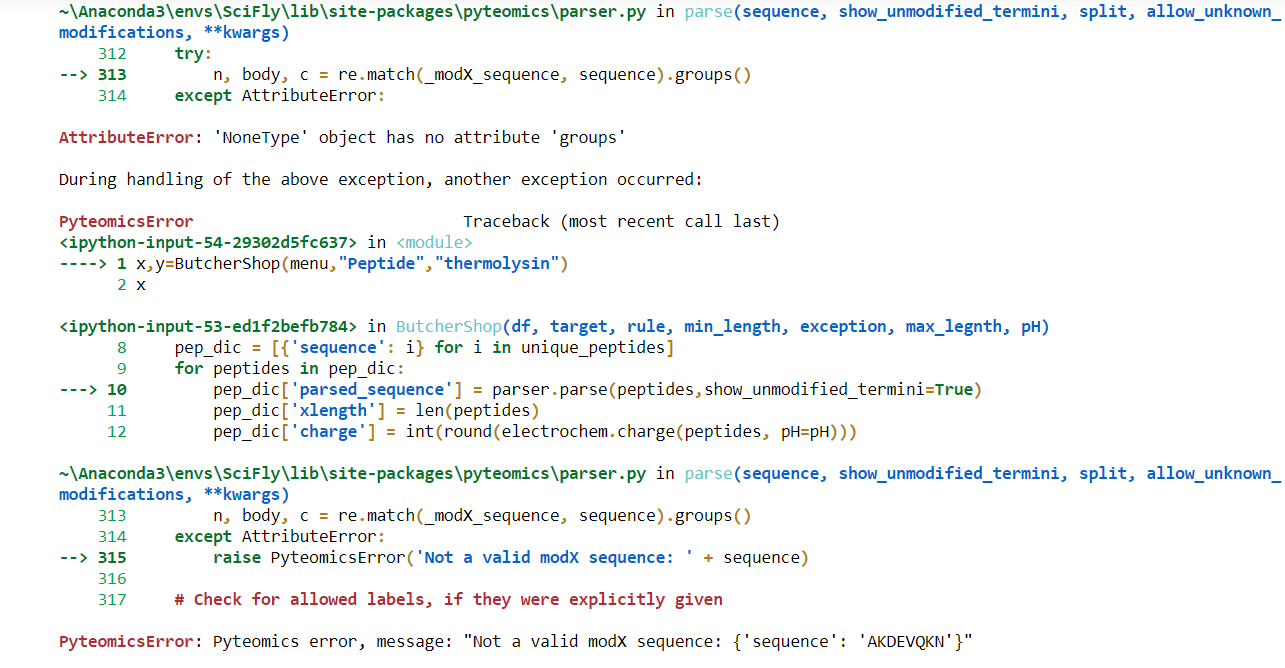
我得到了这个错误:PyteomicsError: Pyteomics错误,消息:"Not序列:{'sequence':'AKDEVQKN'}"
但是,我不确定AKDEVQKN如何与modX_reqquence compilier不匹配:
_modX_sequence = re.compile(r'^([^-]+-)?((?:[^A-Z-]*[A-Z])+)(-[^-]+)?$')根据我对这个正则表达式的理解,它应该找到不以(-)开头的字符串,并且后面是一系列字母字符。
这是我试着使用的函数。
import re
import pyteomics
from pyteomics import fasta, parser
def ButcherShop(df, target, rule,min_length=7,exception=None,max_legnth=100, pH=2.0):
> raw = df[target]
> unique_peptides = set()
> for peptide in raw:
> new_peptides = parser.cleave(peptide, rule=rule,min_length=min_length,exception=exception)
> unique_peptides.update(new_peptides)
> print(f'Done,{len(unique_peptides)} sequences of >= 7 amino acids!')
> pep_dic = [{'sequence': i} for i in unique_peptides]
> for peptides in pep_dic:
> pep_dic['parsed_sequence'] = parser.parse(peptides,show_unmodified_termini=False)
> pep_dic['xlength'] = len(peptides)
> pep_dic['charge'] = int(round(electrochem.charge(peptides, pH=pH)))
> pep_dic['mass']=int(round(Peptide_mass(peptides)))
> pep_dic = [peptide for peptide in pep_dic if peptide['length'] <= int(max_length)]
> pep_df = pd.DataFrame.from_dict(pep_dic)
> return unique_peptides,pep_dic,pep_df感谢您对如何解决这一问题的任何见解。
**更新:如果我在不同的集合上运行,我会得到相同的错误,这可能意味着它是库本身。
错误截图:
回答 3
Stack Overflow用户
发布于 2021-03-07 22:45:46
梨组学保持器在这里。
错误消息实际上告诉您问题的根源:PyteomicsError: Pyteomics error, message: "Not a valid modX sequence: {'sequence': 'AKDEVQKN'}"
这意味着,您传递的不是字符串'AKDEVQKN',而是一个字典{'sequence': 'AKDEVQKN'}。这实际上发生在这里:
pep_dic = [{'sequence': i} for i in unique_peptides]
for peptides in pep_dic:
pep_dic['parsed_sequence'] = parser.parse(peptides,show_unmodified_termini=False)
...您应该将序列本身传递给parse,而不是dict:
pep_dic['parsed_sequence'] = parser.parse(peptides['sequence'], show_unmodified_termini=False)Stack Overflow用户
发布于 2021-03-05 17:18:05
不是解决办法,而是分析..。
在下面的简单示例代码中,'AKDEVQKN‘使用post中的regex进行匹配。
import re
line = 'AKDEVQKN'
pat = re.compile(r'^([^-]+-)?((?:[^A-Z-]*[A-Z])+)(-[^-]+)?$')
x = re.match(pat, line)
if x:
print(x)
print(x.group())
print(x.groups())产出:
<re.Match object; span=(0, 8), match='AKDEVQKN'>
AKDEVQKN
(None, 'AKDEVQKN', None)这表明问题在代码中的其他地方。
- “AKDEVQKN”是完整的行还是有更多的行?
- 是否有可能在使用序列'AKDEVQKN‘调用_modX_sequence时更改了re.match?要检查,请暂时更改
~\Anaconda\envs\SciFly\lib\site-packages\pyteomics\parser.py第312行:
try:
n, body, c = re.match(_modX_sequence, sequence).groups()
except AttributeError: 至
try:
if sequence == 'AKDEVQKN':
print("DEBUG: ", sequence, _modX_sequence)
# or drop into a debugger, pdb or iPython's
# import pdb; pdb.set_trace()
# dir()
n, body, c = re.match(_modX_sequence, sequence).groups()
except AttributeError:Stack Overflow用户
发布于 2021-03-05 17:45:32
在我运行解析器之前,尝试使用它们的有效函数来测试所有的肽。我在绳子上找不到假的。我现在看的是他们的功能或我自己的。
> for peptide in menu["Peptide"]:
> x=parser.valid(peptide)
> if x == False:
> print(peptide)
> break
> else:
> print(x)https://stackoverflow.com/questions/66483290
复制相似问题
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号